proteasome-activating nucleotidase complex / proteasomal protein catabolic process / modification-dependent protein catabolic process / ATP hydrolysis activity / ATP binding Similarity search - Function
Proteasome ATPase / Proteasomal ATPase, N-terminal OB domain / Proteasomal ATPase OB N-terminal domain / Proteasomal ATPase OB C-terminal domain / Proteasomal ATPase OB C-terminal domain / : / ATPase, AAA-type, conserved site / AAA-protein family signature. / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core ...Proteasome ATPase / Proteasomal ATPase, N-terminal OB domain / Proteasomal ATPase OB N-terminal domain / Proteasomal ATPase OB C-terminal domain / Proteasomal ATPase OB C-terminal domain / : / ATPase, AAA-type, conserved site / AAA-protein family signature. / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / Nucleic acid-binding, OB-fold / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
National Institutes of Health/National Center for Complementary and Integrative Health (NIH/NCCIH)
AI070285
United States
National Institutes of Health/National Heart, Lung, and Blood Institute (NIH/NHLBI)
AI088075
United States
Citation
Journal: J Biol Chem / Year: 2021 Title: The mycobacterial proteasomal ATPase Mpa forms a gapped ring to engage the 20S proteasome. Authors: Yanting Yin / Amanda Kovach / Hao-Chi Hsu / K Heran Darwin / Huilin Li / Abstract: Although many bacterial species do not possess proteasome systems, the actinobacteria, including the human pathogen Mycobacterium tuberculosis, use proteasome systems for targeted protein removal. ...Although many bacterial species do not possess proteasome systems, the actinobacteria, including the human pathogen Mycobacterium tuberculosis, use proteasome systems for targeted protein removal. Previous structural analyses of the mycobacterial proteasome ATPase Mpa revealed a general structural conservation with the archaeal proteasome-activating nucleotidase and eukaryotic proteasomal Rpt1-6 ATPases, such as the N-terminal coiled-coil domain, oligosaccharide-/oligonucleotide-binding domain, and ATPase domain. However, Mpa has a unique β-grasp domain that in the ADP-bound crystal structure appears to interfere with the docking to the 20S proteasome core particle (CP). Thus, it is unclear how Mpa binds to proteasome CPs. In this report, we show by cryo-EM that the Mpa hexamer in the presence of a degradation substrate and ATP forms a gapped ring, with two of its six ATPase domains being highly flexible. We found that the linkers between the oligonucleotide-binding and ATPase domains undergo conformational changes that are important for function, revealing a previously unappreciated role of the linker region in ATP hydrolysis-driven protein unfolding. We propose that this gapped ring configuration is an intermediate state that helps rearrange its β-grasp domains and activating C termini to facilitate engagement with proteasome CPs. This work provides new insights into the crucial process of how an ATPase interacts with a bacterial proteasome protease.
History
Deposition
Jan 29, 2021
-
Header (metadata) release
May 5, 2021
-
Map release
May 5, 2021
-
Update
Mar 6, 2024
-
Current status
Mar 6, 2024
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Entire : Cryo-EM structure of the Mpa hexamer in the presence of ATP and t...
Entire
Name: Cryo-EM structure of the Mpa hexamer in the presence of ATP and the Pup-FabD substrate
Components
Complex: Cryo-EM structure of the Mpa hexamer in the presence of ATP and the Pup-FabD substrate
Protein or peptide: AAA ATPase forming ring-shaped complexes
Ligand: MAGNESIUM ION
Ligand: ADENOSINE-5'-TRIPHOSPHATE
Ligand: ADENOSINE-5'-DIPHOSPHATE
-
Supramolecule #1: Cryo-EM structure of the Mpa hexamer in the presence of ATP and t...
Supramolecule
Name: Cryo-EM structure of the Mpa hexamer in the presence of ATP and the Pup-FabD substrate type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Mycobacterium tuberculosis (bacteria)
Mycobacterium tuberculosis (bacteria) Authors
Authors United States, 2 items
United States, 2 items  Citation
Citation Structure visualization
Structure visualization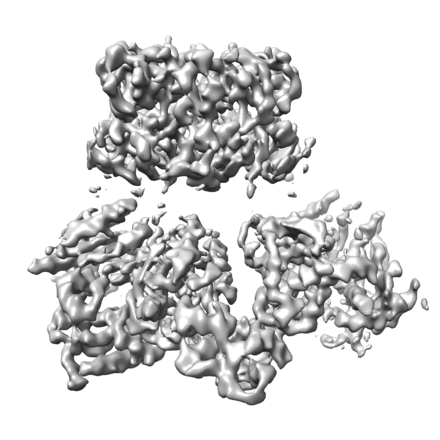
 Downloads & links
Downloads & links emd_23392.png
emd_23392.png http://ftp.pdbj.org/pub/emdb/structures/EMD-23392
http://ftp.pdbj.org/pub/emdb/structures/EMD-23392











 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















 Sample components
Sample components

 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
