Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10351 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
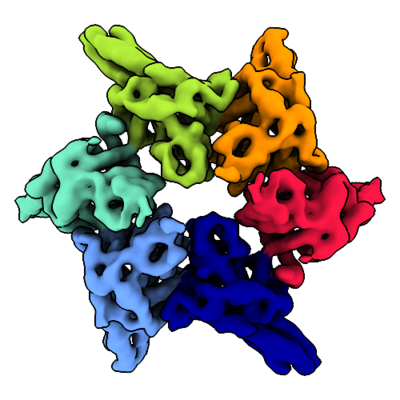
| Title | MoxR AAA-ATPase RavA, C2-symmetric closed ring conformation | |||||||||
 Map data Map data | MoxR AAA-ATPase RavA, C2-symmetric closed ring conformation | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | AAA+ ATPase / MoxR / Escherichia coli / CHAPERONE | |||||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on acid anhydrides; Acting on acid anhydrides to catalyse transmembrane movement of substances / ATP hydrolysis activity / ATP binding / identical protein binding / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
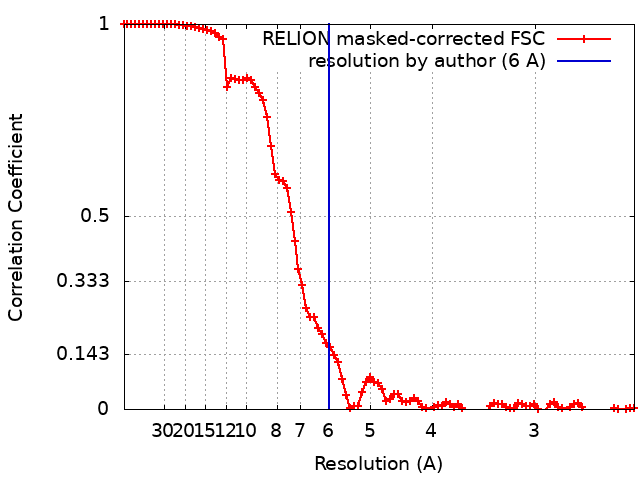
| Method | single particle reconstruction / cryo EM / Resolution: 6.0 Å | |||||||||
 Authors Authors | Jessop M / Felix J | |||||||||
| Funding support | 1 items
| |||||||||
 Citation Citation | Journal: Commun Biol / Year: 2020 Title: Structural insights into ATP hydrolysis by the MoxR ATPase RavA and the LdcI-RavA cage-like complex. Authors: Matthew Jessop / Benoit Arragain / Roger Miras / Angélique Fraudeau / Karine Huard / Maria Bacia-Verloop / Patrice Catty / Jan Felix / Hélène Malet / Irina Gutsche /  Abstract: The hexameric MoxR AAA+ ATPase RavA and the decameric lysine decarboxylase LdcI form a 3.3 MDa cage, proposed to assist assembly of specific respiratory complexes in E. coli. Here, we show that ...The hexameric MoxR AAA+ ATPase RavA and the decameric lysine decarboxylase LdcI form a 3.3 MDa cage, proposed to assist assembly of specific respiratory complexes in E. coli. Here, we show that inside the LdcI-RavA cage, RavA hexamers adopt an asymmetric spiral conformation in which the nucleotide-free seam is constrained to two opposite orientations. Cryo-EM reconstructions of free RavA reveal two co-existing structural states: an asymmetric spiral, and a flat C2-symmetric closed ring characterised by two nucleotide-free seams. The closed ring RavA state bears close structural similarity to the pseudo two-fold symmetric crystal structure of the AAA+ unfoldase ClpX, suggesting a common ATPase mechanism. Based on these structures, and in light of the current knowledge regarding AAA+ ATPases, we propose different scenarios for the ATP hydrolysis cycle of free RavA and the LdcI-RavA cage-like complex, and extend the comparison to other AAA+ ATPases of clade 7. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10351.map.gz | 3.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10351-v30.xmlemd-10351.xml | 10.8 KB 10.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_10351_fsc.xml | 9.2 KB | Display | FSC data file |
| Images |  emd_10351.png emd_10351.png | 143.8 KB | ||
| Filedesc metadata | emd-10351.cif.gz | 5.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10351ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10351 http://ftp.pdbj.org/pub/emdb/structures/EMD-10351ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10351 | HTTPS FTP |
-Validation report
| Summary document | emd_10351_validation.pdf.gz | 364.3 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_10351_full_validation.pdf.gz | 363.9 KB | Display | |
| Data in XML | emd_10351_validation.xml.gz | 11.1 KB | Display | |
| Data in CIF | emd_10351_validation.cif.gz | 14.5 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-10351ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-10351 | HTTPS FTP |
-Related structure data
| Related structure data |  6szaMC  4469C  4470C  6q7lC  6q7mC  6szbC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_10351.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | MoxR AAA-ATPase RavA, C2-symmetric closed ring conformation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.21 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : RavA + Mg-ADP
| Entire | Name: RavA + Mg-ADP |
|---|---|
| Components |
|
-Supramolecule #1: RavA + Mg-ADP
| Supramolecule | Name: RavA + Mg-ADP / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 336 KDa |
-Macromolecule #1: RavA
| Macromolecule | Name: RavA / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 56.454586 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MAHPHLLAER ISRLSSSLEK GLYERSHAIR LCLLAALSGE SVFLLGPPGI AKSLIARRLK FAFQNARAFE YLMTRFSTPE EVFGPLSIQ ALKDEGRYER LTSGYLPEAE IVFLDEIWKA GPAILNTLLT AINERQFRNG AHVEKIPMRL LVAASNELPE A DSSLEALY ...String: MAHPHLLAER ISRLSSSLEK GLYERSHAIR LCLLAALSGE SVFLLGPPGI AKSLIARRLK FAFQNARAFE YLMTRFSTPE EVFGPLSIQ ALKDEGRYER LTSGYLPEAE IVFLDEIWKA GPAILNTLLT AINERQFRNG AHVEKIPMRL LVAASNELPE A DSSLEALY DRMLIRLWLD KVQDKANFRS MLTSQQDEND NPVPDALQVT DEEYERWQKE IGEITLPDHV FELIFMLRQQ LD KLPDAPY VSDRRWKKAI RLLQASAFFS GRSAVAPVDL ILLKDCLWYD AQSLNLIQQQ IDVLMTGHAW QQQGMLTRLG AIV QRHLQL QQQQSDKTAL TVIRLGGIFS RRQQYQLPVN VTASTLTLLL QKPLKLHDME VVHISFERSA LEQWLSKGGE IRGK LNGIG FAQKLNLEVD SAQHLVVRDV SLQGSTLALP GSSAEGLPGE IKQQLEELES DWRKQHALFS EQQKCLFIPG DWLGR IEAS LQDVGAQIRQ AQQC UniProtKB: ATPase RavA |
-Macromolecule #2: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 2 / Number of copies: 4 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |