Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-9233 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
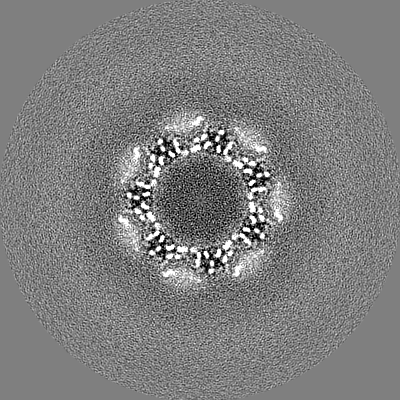
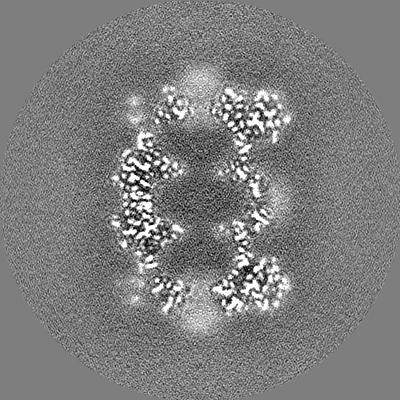
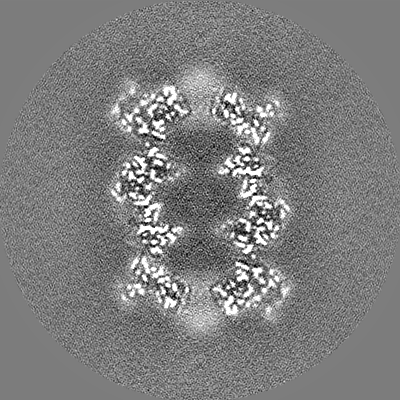
| Title | T20S proteasome | |||||||||
 Map data Map data | T20S sharpened masked map | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Thermoplasma acidophilum (acidophilic) Thermoplasma acidophilum (acidophilic) | |||||||||
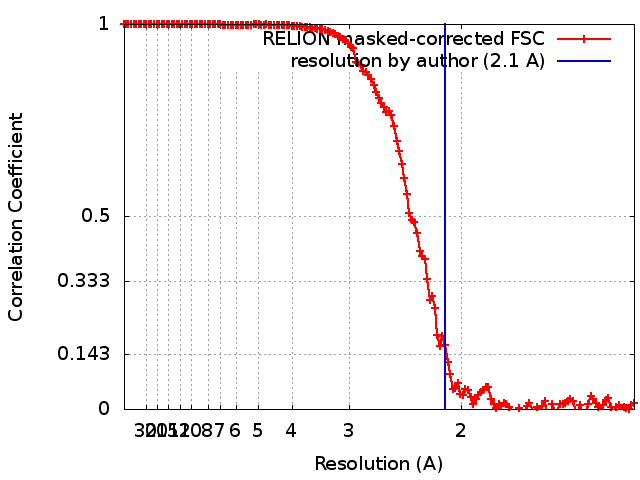
| Method | single particle reconstruction / cryo EM / Resolution: 2.1 Å | |||||||||
 Authors Authors | Eng ET / Kopylov M / Jordan KJ / Rice WJ / Carragher BO / Potter CS | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2019 Title: Reducing cryoEM file storage using lossy image formats. Authors: Edward T Eng / Mykhailo Kopylov / Carl J Negro / Sarkis Dallaykan / William J Rice / Kelsey D Jordan / Kotaro Kelley / Bridget Carragher / Clinton S Potter /  Abstract: Recent advances in instrumentation and software for cryoEM have increased the applicability and utility of this method. High levels of automation and faster data acquisition rates require hard ...Recent advances in instrumentation and software for cryoEM have increased the applicability and utility of this method. High levels of automation and faster data acquisition rates require hard decisions to be made regarding data retention. Here we investigate the efficacy of data compression applied to aligned summed movie files. Surprisingly, these images can be compressed using a standard lossy method that reduces file storage by 90-95% and yet can still be processed to provide sub-2 Å reconstructed maps. We do not advocate this as an archival method, but it may provide a useful means for retaining images as an historical record, especially at large facilities. | |||||||||
| History |
|
- Structure visualization
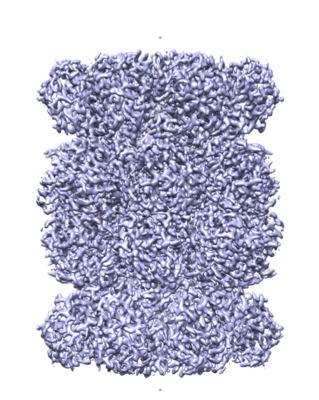
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_9233.map.gz | 30.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-9233-v30.xmlemd-9233.xml | 19.8 KB 19.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_9233_fsc.xml | 14.1 KB | Display | FSC data file |
| Images |  emd_9233.png emd_9233.png | 217.7 KB | ||
| Masks | emd_9233_msk_1.map | 244.1 MB | Mask map | |
| Others | emd_9233_additional.map.gzemd_9233_half_map_1.map.gzemd_9233_half_map_2.map.gz | 192.9 MB 194 MB 194.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-9233ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9233 http://ftp.pdbj.org/pub/emdb/structures/EMD-9233ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9233 | HTTPS FTP |
-Related structure data
| Related structure data |  9203C C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10218 (Title: Thermoplasma acidophilum 20S / Data size: 1.5 TB Data #1: unaligned multi-frame micrographs of T20S (18mar31a) [micrographs - multiframe]) |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_9233.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | T20S sharpened masked map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.6616 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
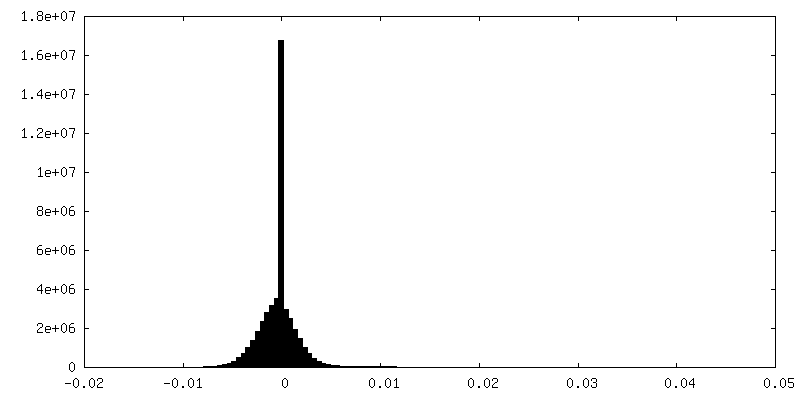
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)












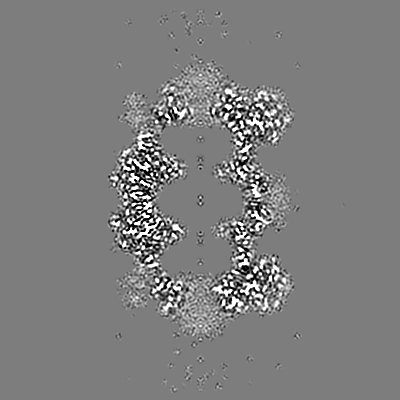







-Supplemental data
-Mask #1
| File | emd_9233_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
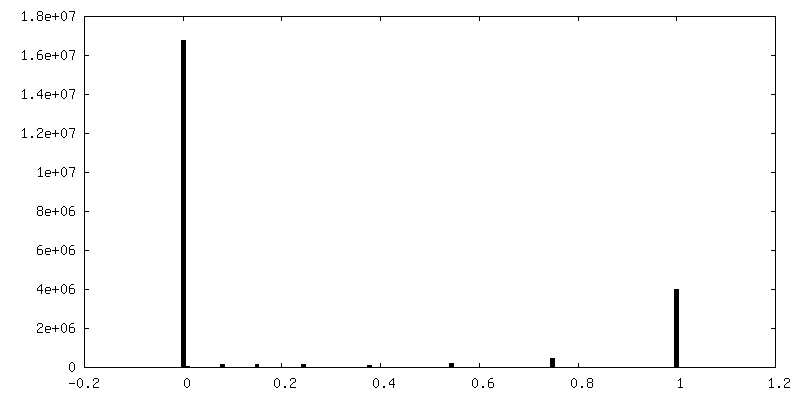
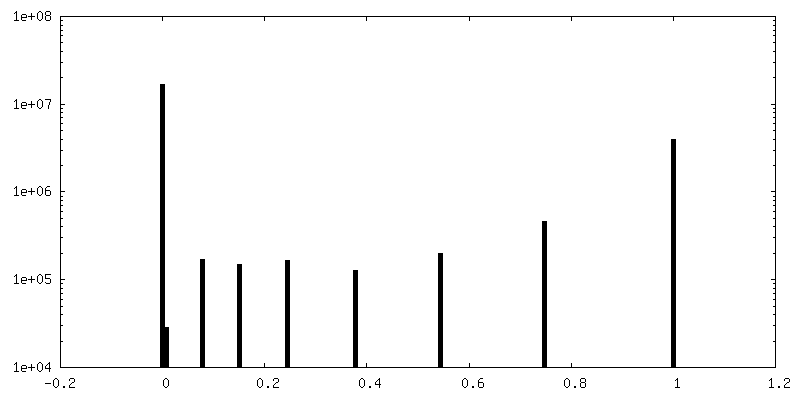
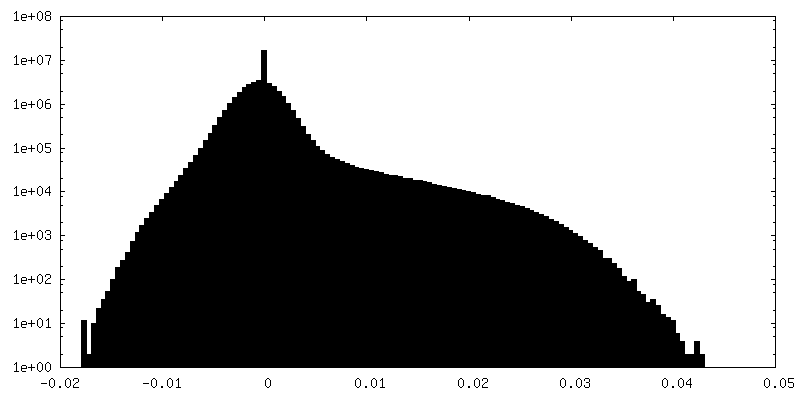
| Density Histograms |






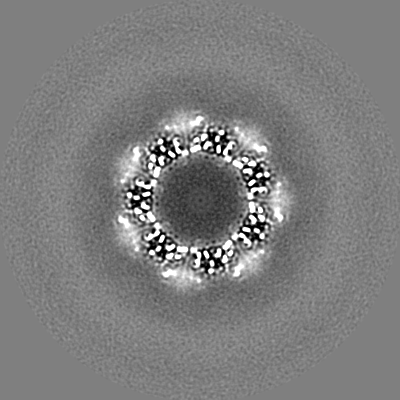
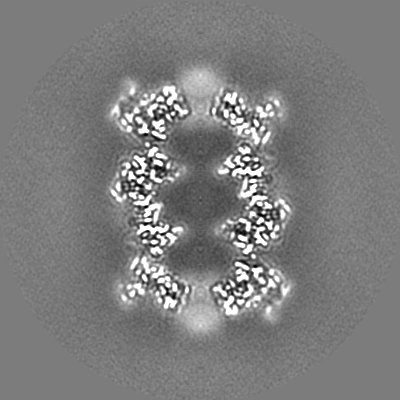
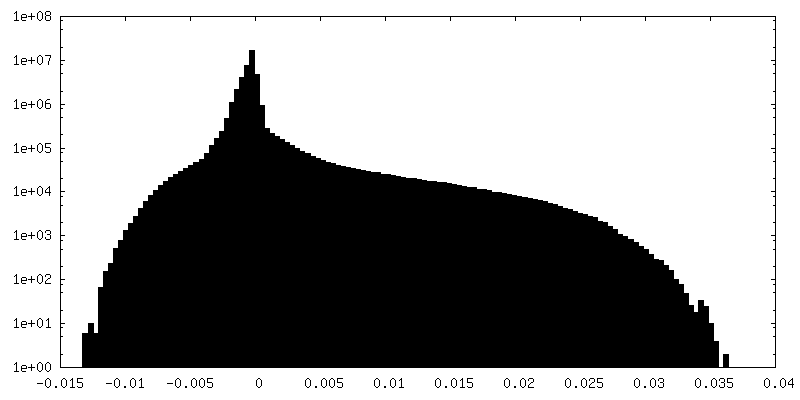
-Additional map: T20S unsharpened map
| File | emd_9233_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | T20S unsharpened map | ||||||||||||
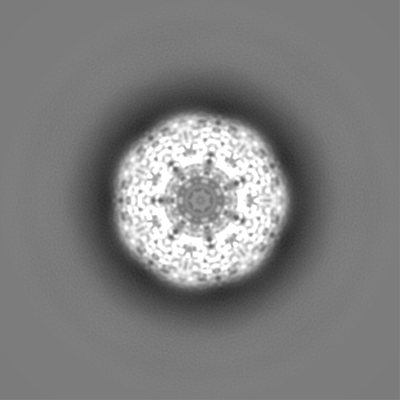
| Projections & Slices |
| ||||||||||||
| Density Histograms |




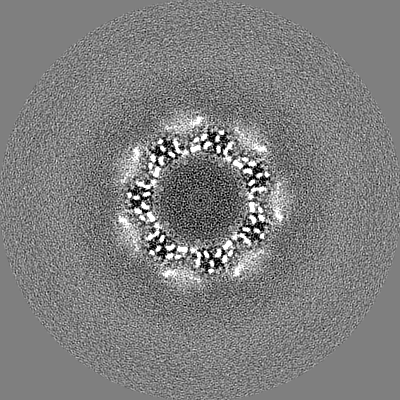
-Half map: T20S even half map
| File | emd_9233_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
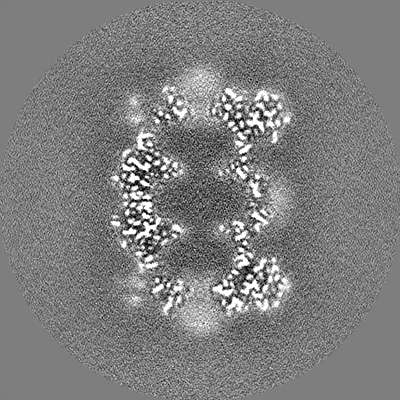
| Annotation | T20S even half map | ||||||||||||
| Projections & Slices |
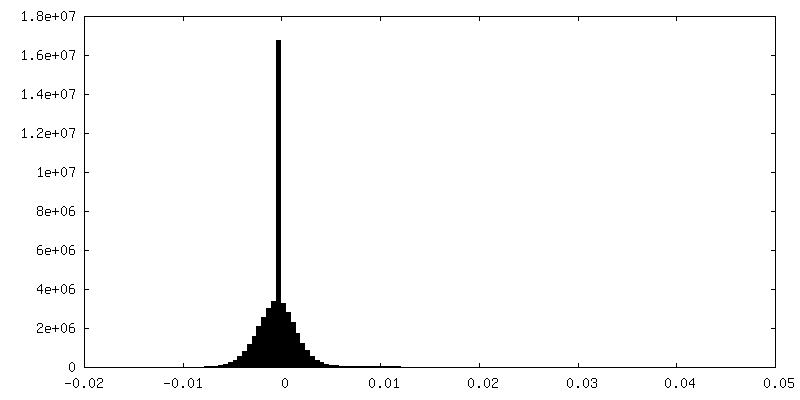
| ||||||||||||
| Density Histograms |



-Half map: T20S odd half map
| File | emd_9233_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
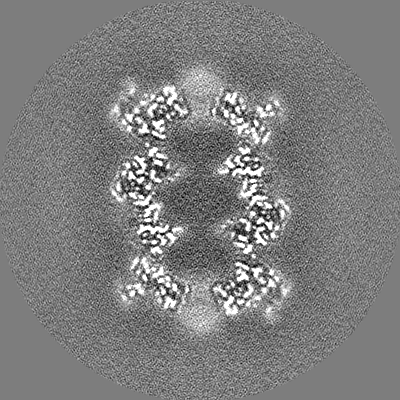
| Annotation | T20S odd half map | ||||||||||||
| Projections & Slices |
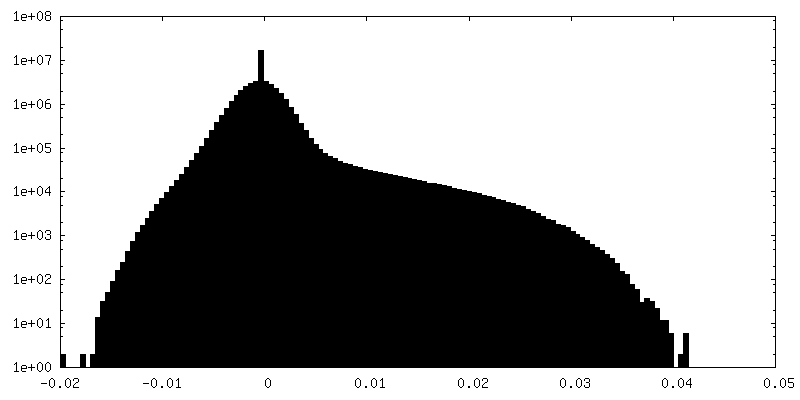
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : 20S proteasome
| Entire | Name: 20S proteasome |
|---|---|
| Components |
|
-Supramolecule #1: 20S proteasome
| Supramolecule | Name: 20S proteasome / type: complex / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Thermoplasma acidophilum (acidophilic) |
| Recombinant expression | Organism:  |
| Molecular weight | Theoretical: 700 KDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.21 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
Details: 20 mM Tris, 150 mM NaCl | |||||||||
| Grid | Model: C-flat-1.2/1.3 4C / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: OTHER | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 75 % / Chamber temperature: 298 K / Instrument: GATAN CRYOPLUNGE 3 Details: Blotted for 2.5 seconds after a 30 second wait time.. | |||||||||
| Details | 1.2/1.3 C-Flat grid, plasma cleaned |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Details | Leginon |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-50 / Number grids imaged: 1 / Number real images: 1173 / Average exposure time: 6.259 sec. / Average electron dose: 62.59 e/Å2 Details: Images were collected in movie mode at 5 frames per second. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 3.02 µm / Calibrated defocus min: 0.2 µm / Calibrated magnification: 75574 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 37000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |