Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-4260: Volta phase plate data collection facilitates image processing an... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4260 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Volta phase plate data collection facilitates image processing and cryo-EM structure determination | |||||||||
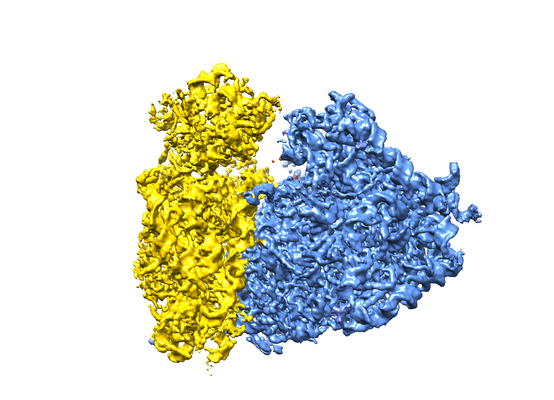
 Map data Map data | human 80S ribosome reconstructed from close to focus VPP data, postprocessed map. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
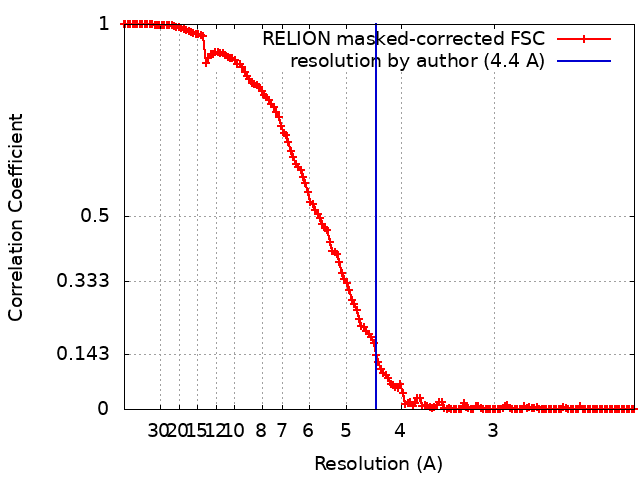
| Method | single particle reconstruction / cryo EM / Resolution: 4.4 Å | |||||||||
 Authors Authors | von Loeffelholz O / Klaholz BP / Natchiar SK | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2018 Title: Volta phase plate data collection facilitates image processing and cryo-EM structure determination. Authors: Ottilie von Loeffelholz / Gabor Papai / Radostin Danev / Alexander G Myasnikov / S Kundhavai Natchiar / Isabelle Hazemann / Jean-François Ménétret / Bruno P Klaholz /   Abstract: A current bottleneck in structure determination of macromolecular complexes by cryo electron microscopy (cryo-EM) is the large amount of data needed to obtain high-resolution 3D reconstructions, ...A current bottleneck in structure determination of macromolecular complexes by cryo electron microscopy (cryo-EM) is the large amount of data needed to obtain high-resolution 3D reconstructions, including through sorting into different conformations and compositions with advanced image processing. Additionally, it may be difficult to visualize small ligands that bind in sub-stoichiometric levels. Volta phase plates (VPP) introduce a phase shift in the contrast transfer and drastically increase the contrast of the recorded low-dose cryo-EM images while preserving high frequency information. Here we present a comparative study to address the behavior of different data sets during image processing and quantify important parameters during structure refinement. The automated data collection was done from the same human ribosome sample either as a conventional defocus range dataset or with a Volta phase plate close to focus (cfVPP) or with a small defocus (dfVPP). The analysis of image processing parameters shows that dfVPP data behave more robustly during cryo-EM structure refinement because particle alignments, Euler angle assignments and 2D & 3D classifications behave more stably and converge faster. In particular, less particle images are required to reach the same resolution in the 3D reconstructions. Finally, we find that defocus range data collection is also applicable to VPP. This study shows that data processing and cryo-EM map interpretation, including atomic model refinement, are facilitated significantly by performing VPP cryo-EM, which will have an important impact on structural biology. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4260.map.gz | 264.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4260-v30.xmlemd-4260.xml | 9.2 KB 9.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_4260_fsc.xml | 14.5 KB | Display | FSC data file |
| Images | 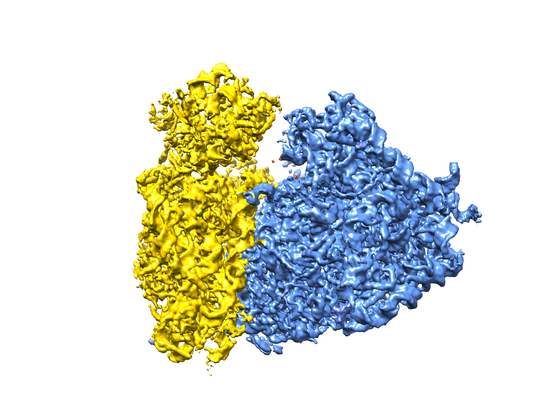 emd_4260.png emd_4260.png | 182.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4260ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4260 http://ftp.pdbj.org/pub/emdb/structures/EMD-4260ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4260 | HTTPS FTP |
-Validation report
| Summary document | emd_4260_validation.pdf.gz | 357.8 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_4260_full_validation.pdf.gz | 356.9 KB | Display | |
| Data in XML | emd_4260_validation.xml.gz | 14.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-4260ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-4260 | HTTPS FTP |
-Related structure data
| Related structure data |  4261C  4262C C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10154 (Title: Volta phase plate data collection facilitates image processing and cryo-EM structure determination Data size: 89.4 Data #1: Unaligned multi-frame micrographs of human ribosome, collected with VPP close to focus (cfVPP) [micrographs - multiframe]) |












-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_4260.map.gz / Format: CCP4 / Size: 282.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | human 80S ribosome reconstructed from close to focus VPP data, postprocessed map. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.09 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : ribosome
| Entire | Name: ribosome |
|---|---|
| Components |
|
-Supramolecule #1: ribosome
| Supramolecule | Name: ribosome / type: complex / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / Strain: HeLa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.6 |
|---|---|
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN |
|---|---|
| Specialist optics | Phase plate: VOLTA PHASE PLATE Spherical aberration corrector: CS corrector used, opening up to 13 mrad at 26 mrad tilt Energy filter - Name: GIF Quantum LS |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 7420 pixel / Digitization - Dimensions - Height: 7676 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-29 / Number grids imaged: 1 / Number real images: 433 / Average exposure time: 4.5 sec. / Average electron dose: 1.03 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 45871 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 0.01 mm / Nominal defocus max: 0.05 µm / Nominal defocus min: 0.05 µm / Nominal magnification: 45871 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |