 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
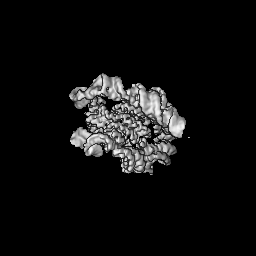
| Title | Native Tetrahymena ribozyme conformation | ||||||||||||||||||
 Map data Map data | Cryo-EM structure of native Tetrahymena ribozyme conformation --- unsharpen | ||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | Misfolded Tetrahymena Ribozyme / Topological Crossing / Cryo-EM / refolding / RNA | ||||||||||||||||||
| Biological species |   Tetrahymena thermophila (eukaryote) Tetrahymena thermophila (eukaryote) | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.01 Å | ||||||||||||||||||
 Authors Authors | Li S / Palo M / Pintilie G / Zhang X / Su Z / Kappel K / Chiu W / Zhang K / Das R | ||||||||||||||||||
| Funding support |  United States, 5 items United States, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2022 Title: Topological crossing in the misfolded ribozyme resolved by cryo-EM. Authors: Shanshan Li / Michael Z Palo / Grigore Pintilie / Xiaojing Zhang / Zhaoming Su / Kalli Kappel / Wah Chiu / Kaiming Zhang / Rhiju Das /  Abstract: The group I intron has been a key system in the understanding of RNA folding and misfolding. The molecule folds into a long-lived misfolded intermediate (M) , which has been known to form extensive ...The group I intron has been a key system in the understanding of RNA folding and misfolding. The molecule folds into a long-lived misfolded intermediate (M) , which has been known to form extensive native-like secondary and tertiary structures but is separated by an unknown kinetic barrier from the native state (N). Here, we used cryogenic electron microscopy (cryo-EM) to resolve misfolded structures of the L-21 ScaI ribozyme. Maps of three M substates (M1, M2, M3) and one N state were achieved from a single specimen with overall resolutions of 3.5 Å, 3.8 Å, 4.0 Å, and 3.0 Å, respectively. Comparisons of the structures reveal that all the M substates are highly similar to N, except for rotation of a core helix P7 that harbors the ribozyme's guanosine binding site and the crossing of the strands J7/3 and J8/7 that connect P7 to the other elements in the ribozyme core. This topological difference between the M substates and N state explains the failure of 5'-splice site substrate docking in M, supports a topological isomer model for the slow refolding of M to N due to a trapped strand crossing, and suggests pathways for M-to-N refolding. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_33428.map.gz | 31.8 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-33428-v30.xmlemd-33428.xml | 21.4 KB 21.4 KB | Display Display | EMDB header |
| Images | 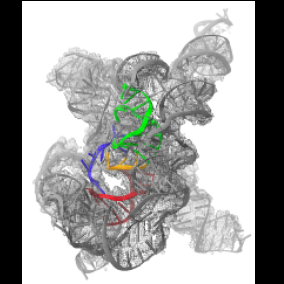 emd_33428.png emd_33428.png | 55.9 KB | ||
| Filedesc metadata | emd-33428.cif.gz | 5.7 KB | ||
| Others | emd_33428_additional_1.map.gzemd_33428_half_map_1.map.gzemd_33428_half_map_2.map.gz | 59.8 MB 59.4 MB 59.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-33428ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33428 http://ftp.pdbj.org/pub/emdb/structures/EMD-33428ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33428 | HTTPS FTP |
-Related structure data
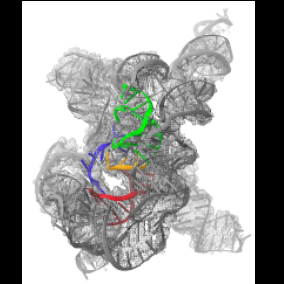
| Related structure data |  7xsnMC  7xskC  7xslC  7xsmC M: atomic model generated by this map C: citing same article ( |
|---|



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_33428.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM structure of native Tetrahymena ribozyme conformation --- unsharpen | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.82 Å | ||||||||||||||||||||||||||||||||||||
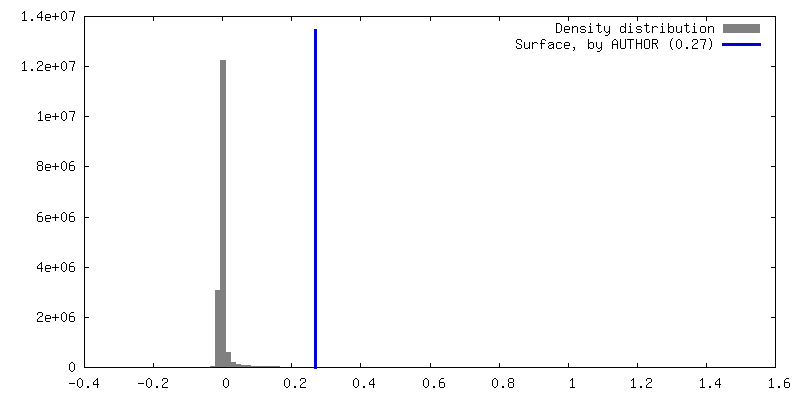
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)















-Supplemental data
-Additional map: Cryo-EM structure of native Tetrahymena ribozyme conformation ---...
| File | emd_33428_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
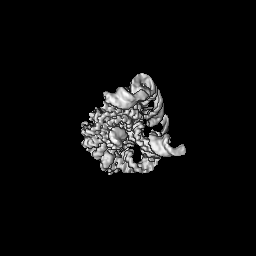
| Annotation | Cryo-EM structure of native Tetrahymena ribozyme conformation --- sharpen | ||||||||||||
| Projections & Slices |
| ||||||||||||
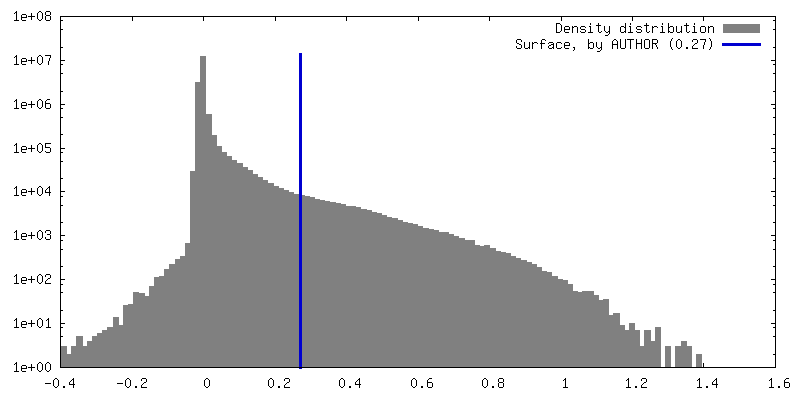
| Density Histograms |






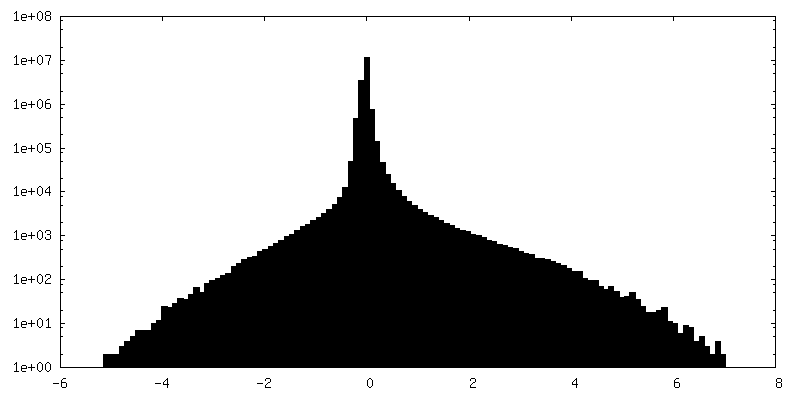
-Half map: Half map 1
| File | emd_33428_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
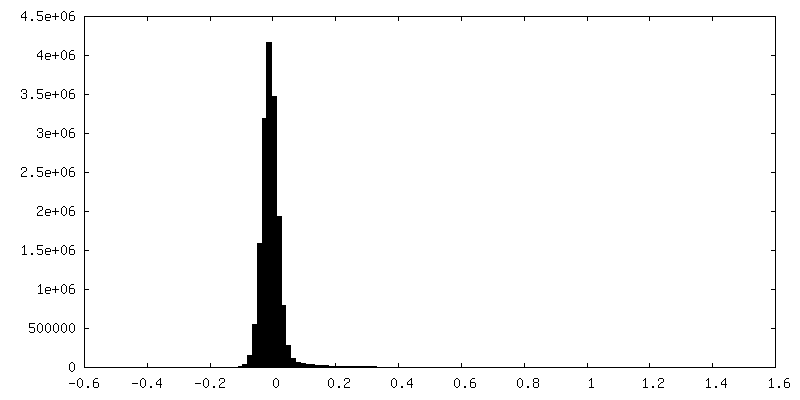
| Density Histograms |



-Half map: Half map 2
| File | emd_33428_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
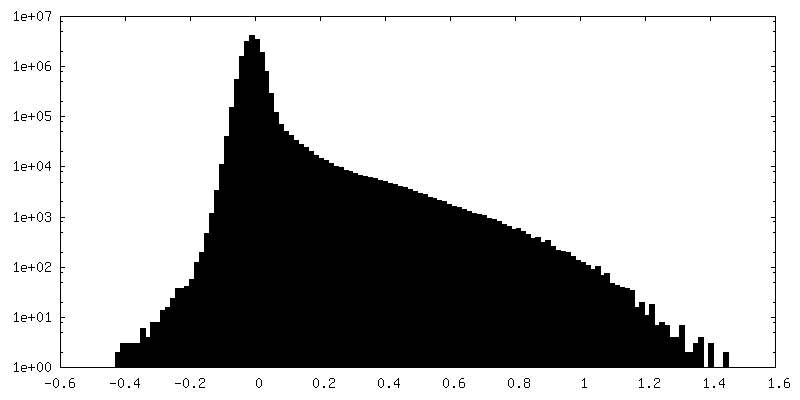
| Density Histograms |





- Sample components
Sample components
-Entire : Native Tetrahymena ribozyme conformation
| Entire | Name: Native Tetrahymena ribozyme conformation |
|---|---|
| Components |
|
-Supramolecule #1: Native Tetrahymena ribozyme conformation
| Supramolecule | Name: Native Tetrahymena ribozyme conformation / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Tetrahymena thermophila (eukaryote) |
| Molecular weight | Theoretical: 120 KDa |
-Macromolecule #1: RNA (387-MER)
| Macromolecule | Name: RNA (387-MER) / type: rna / ID: 1 / Number of copies: 1 |
|---|---|
| Source (natural) | Organism: Tetrahymena thermophila (eukaryote) |
| Molecular weight | Theoretical: 125.402945 KDa |
| Sequence | String: GGAGGGAAAA GUUAUCAGGC AUGCACCUGG UAGCUAGUCU UUAAACCAAU AGAUUGCAUC GGUUUAAAAG GCAAGACCGU CAAAUUGCG GGAAAGGGGU CAACAGCCGU UCAGUACCAA GUCUCAGGGG AAACUUUGAG AUGGCCUUGC AAAGGGUAUG G UAAUAAGC ...String: GGAGGGAAAA GUUAUCAGGC AUGCACCUGG UAGCUAGUCU UUAAACCAAU AGAUUGCAUC GGUUUAAAAG GCAAGACCGU CAAAUUGCG GGAAAGGGGU CAACAGCCGU UCAGUACCAA GUCUCAGGGG AAACUUUGAG AUGGCCUUGC AAAGGGUAUG G UAAUAAGC UGACGGACAU GGUCCUAACC ACGCAGCCAA GUCCUAAGUC AACAGAUCUU CUGUUGAUAU GGAUGCAGUU CA CAGACUA AAUGUCGGUC GGGGAAGAUG UAUUCUUCUC AUAAGAUAUA GUCGGACCUC UCCUUAAUGG GAGCUAGCGG AUG AAGUGA UGCAACACUG GAGCCGCUGG GAACUAAUUU GUAUGCGAAA GUAUAUUGAU UAGUUUUGGA GU GENBANK: GENBANK: X54512.1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 8 |
|---|---|
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Number real images: 42382 / Average electron dose: 51.3 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.8000000000000003 µm / Nominal defocus min: 1.2 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
