 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1asm: CRYSTAL STRUCTURES OF ESCHERICHIA COLI ASPARTATE AMINOTRANSFERASE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1asm | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURES OF ESCHERICHIA COLI ASPARTATE AMINOTRANSFERASE IN TWO CONFORMATIONS: COMPARISON OF AN UNLIGANDED OPEN AND TWO LIGANDED CLOSED FORMS | ||||||
 Components Components | ASPARTATE AMINOTRANSFERASE | ||||||
 Keywords Keywords | AMINOTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology information: / L-tyrosine:2-oxoglutarate transaminase activity / L-phenylalanine biosynthetic process / aspartate transaminase / L-aspartate:2-oxoglutarate transaminase activity / pyridoxal phosphate binding / protein homodimerization activity / identical protein binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.35 Å X-RAY DIFFRACTION / Resolution: 2.35 Å | ||||||
 Authors Authors | Jaeger, J. / Jansonius, J.N. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1994 Title: Crystal structures of Escherichia coli aspartate aminotransferase in two conformations. Comparison of an unliganded open and two liganded closed forms. Authors: Jager, J. / Moser, M. / Sauder, U. / Jansonius, J.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1asm.cif.gz | 166.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1asm.ent.gz | 132.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1asm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/as/1asmftp://data.pdbj.org/pub/pdb/validation_reports/as/1asm | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES PRO 138 AND PRO 195 OF BOTH CHAINS ARE CIS PROLINES. | ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.99999, -0.0043, -0.00157), Vector: Details | THE ASYMMETRIC UNIT CONTAINS A DIMER OF ASPARTATE AMINOTRANSFERASE. | |
-Components
| #1: Protein | Mass: 43619.215 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical |   Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 247.142 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10NO6P#3: Chemical | 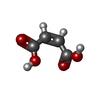  Mass: 116.072 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H4O4 Mass: 116.072 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H4O4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 223 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 223 / Source method: isolated from a natural source / Formula: H2OCompound details | IN BOTH CHAINS, NZ OF LYS 258 FORMS A (PROTONATED | Sequence details | THE RESIDUE NUMBERING USED IN THIS ENTRY CONFORMS TO THE AMINO ACID SEQUENCE OF THE CHICKEN ...THE RESIDUE NUMBERING USED IN THIS ENTRY CONFORMS TO THE AMINO ACID SEQUENCE OF THE CHICKEN CYTOSOLIC ISOENZYME. HENCE, THE RESIDUE NUMBERING STARTS WITH MET 5 AND RESIDUES 127, 128, 130, 131, 132, 153, 232 AND 406 OF BOTH CHAINS HAVE NOT BEEN INCLUDED. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.03 Å3/Da / Density % sol: 59.45 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: referred to 'Smith, D. L.', (1986) J. Mol. Biol., 191, 301-302 | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.35 Å / Num. all: 75087 / Num. obs: 39857 / % possible obs: 91 % / Rmerge(I) obs: 0.044 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.35→10 Å /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.35→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.1878 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 34 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
