 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7zy0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of compound 7 bound to CK2alpha | |||||||||
 Components Components | Casein kinase II subunit alpha | |||||||||
 Keywords Keywords | TRANSFERASE / Fragment based drug discovery | |||||||||
| Function / homology |  Function and homology information Function and homology informationPhosphorylation and nuclear translocation of BMAL1 (ARNTL) and CLOCK / positive regulation of aggrephagy / regulation of chromosome separation / WNT mediated activation of DVL / Condensation of Prometaphase Chromosomes / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / Regulation of CDH1 posttranslational processing and trafficking to plasma membrane / Receptor Mediated Mitophagy ...Phosphorylation and nuclear translocation of BMAL1 (ARNTL) and CLOCK / positive regulation of aggrephagy / regulation of chromosome separation / WNT mediated activation of DVL / Condensation of Prometaphase Chromosomes / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / Regulation of CDH1 posttranslational processing and trafficking to plasma membrane / Receptor Mediated Mitophagy / Sin3-type complex / Synthesis of PC / negative regulation of signal transduction by p53 class mediator / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / Maturation of hRSV A proteins / negative regulation of apoptotic signaling pathway / negative regulation of double-strand break repair via homologous recombination / positive regulation of Wnt signaling pathway / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / Signal transduction by L1 / Hsp90 protein binding / PML body / Wnt signaling pathway / Regulation of PTEN stability and activity / kinase activity / positive regulation of protein catabolic process / KEAP1-NFE2L2 pathway / rhythmic process / double-strand break repair / Cooperation of PDCL (PhLP1) and TRiC/CCT in G-protein beta folding / positive regulation of cell growth / protein folding / Regulation of TP53 Activity through Phosphorylation / non-specific serine/threonine protein kinase / regulation of cell cycle / negative regulation of translation / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / positive regulation of cell population proliferation / apoptotic process / DNA damage response / signal transduction / nucleoplasm / ATP binding / identical protein binding / nucleus / plasma membrane / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.44 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.44 Å | |||||||||
 Authors Authors | Brear, P. / Fusco, C. / Atkinson, E. / Rossmann, M. / Francis, N. / Iegre, J. / Hyvonen, M. / Spring, D. | |||||||||
| Funding support |  United Kingdom, 2items United Kingdom, 2items
| |||||||||
 Citation Citation | Journal: Rsc Med Chem / Year: 2022 Title: A fragment-based approach leading to the discovery of inhibitors of CK2 alpha with a novel mechanism of action. Authors: Brear, P. / De Fusco, C. / Atkinson, E.L. / Iegre, J. / Francis-Newton, N.J. / Venkitaraman, A.R. / Hyvonen, M. / Spring, D.R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7zy0.cif.gz | 89 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7zy0.ent.gz | 64.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7zy0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zy/7zy0ftp://data.pdbj.org/pub/pdb/validation_reports/zy/7zy0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7zy2C  7zy5C  7zy8C  7zydC  7zykC  7zyoC  7zyrC  8ae7C  8aecC  8aekC  8aemC  5mohS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39031.391 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CSNK2A1, CK2A1 / Production host:  References: UniProt: P68400, non-specific serine/threonine protein kinase |
|---|---|
| #2: Chemical | ChemComp-KC0 / 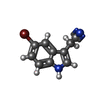  Mass: 235.080 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H7BrN2 / Feature type: SUBJECT OF INVESTIGATION Mass: 235.080 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H7BrN2 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 165 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.03 Å3/Da / Density % sol: 39.32 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 112.5mM Mes pH 6.5, 35% glycerol ethoxylate, 180 mM ammonium acetate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04 / Wavelength: 0.9323 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Mar 8, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9323 Å / Relative weight: 1 |
| Reflection | Resolution: 1.44→58.85 Å / Num. obs: 56651 / % possible obs: 99.6 % / Redundancy: 3.3 % / Biso Wilson estimate: 18.64 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.042 / Rpim(I) all: 0.027 / Rrim(I) all: 0.051 / Rsym value: 0.042 / Net I/σ(I): 14 |
| Reflection shell | Resolution: 1.44→1.444 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.448 / Mean I/σ(I) obs: 2.1 / Num. measured all: 43015 / Num. unique obs: 13499 / CC1/2: 0.838 / Rpim(I) all: 0.247 / Rrim(I) all: 0.451 / Rsym value: 0.448 / Net I/σ(I) obs: 2.8 / % possible all: 99.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5MOH Resolution: 1.44→58.85 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.9271 / SU R Cruickshank DPI: 0.074 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.073 / SU Rfree Blow DPI: 0.07 / SU Rfree Cruickshank DPI: 0.07
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 98.42 Å2 / Biso mean: 22.94 Å2 / Biso min: 8.74 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.165 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.44→58.85 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.44→1.48 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
