- PDB-7oet: C-TERMINAL BROMODOMAIN OF HUMAN BRD2 WITH 1,5-dimethyl-N-(2-(meth... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 7oet
Title
C-TERMINAL BROMODOMAIN OF HUMAN BRD2 WITH 1,5-dimethyl-N-(2-(methylamino)-2-oxo-1-(tetrahydro-2H-pyran-4-yl)ethyl)-6-oxo-N-(2-phenyl-2-(pyridin-2-yl)ethyl)-1,6-dihydropyridine-3-carboxamide
Components
Bromodomain-containing protein 2
Keywords
NUCLEAR PROTEIN / INHIBITOR / HISTONE / EPIGENETIC READER / BROMODOMAIN / BRD2 / BROMODOMAIN CONTAINING PROTEIN 2 / ANTAGONIST
Function / homology
Function and homology information
acetylation-dependent protein binding / chromatin looping / RUNX3 regulates p14-ARF / positive regulation of T-helper 17 cell lineage commitment / protein localization to chromatin / neural tube closure / lysine-acetylated histone binding / nucleosome assembly / spermatogenesis / nuclear speck ...acetylation-dependent protein binding / chromatin looping / RUNX3 regulates p14-ARF / positive regulation of T-helper 17 cell lineage commitment / protein localization to chromatin / neural tube closure / lysine-acetylated histone binding / nucleosome assembly / spermatogenesis / nuclear speck / protein serine/threonine kinase activity / chromatin binding / chromatin / regulation of transcription by RNA polymerase II / nucleoplasm / nucleus / cytoplasm Similarity search - Function
NET domain superfamily / NET domain profile. / Brdt, bromodomain, repeat II / Brdt, bromodomain, repeat I / : / NET domain / Bromodomain extra-terminal - transcription regulation / Bromodomain, conserved site / Bromodomain signature. / Bromodomain ...NET domain superfamily / NET domain profile. / Brdt, bromodomain, repeat II / Brdt, bromodomain, repeat I / : / NET domain / Bromodomain extra-terminal - transcription regulation / Bromodomain, conserved site / Bromodomain signature. / Bromodomain / Bromodomain profile. / bromo domain / Bromodomain / Bromodomain-like superfamily Similarity search - Domain/homology
Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Feb 15, 2018
Radiation
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.966 Å / Relative weight: 1
Reflection
Resolution: 1.41→42.29 Å / Num. obs: 23909 / % possible obs: 99.3 % / Redundancy: 4.3 % / CC1/2: 0.999 / Rmerge(I) obs: 0.037 / Net I/σ(I): 17.4
Reflection shell
Resolution: 1.41→1.43 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.656 / Mean I/σ(I) obs: 2.1 / Num. unique obs: 1157 / CC1/2: 0.718 / % possible all: 99.6
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0258
refinement
XDS
datareduction
Aimless
datascaling
Refinement
Method to determine structure: FOURIER SYNTHESIS Starting model: in house structure Resolution: 1.41→15.696 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.964 / SU B: 1.217 / SU ML: 0.046 / Cross valid method: FREE R-VALUE / ESU R: 0.063 / ESU R Free: 0.062 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Rfree
0.1959
1204
5.074 %
Rwork
0.18
22527
-
all
0.181
-
-
obs
-
23731
99.226 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL PLUS MASK
Displacement parameters
Biso mean: 28.07 Å2
Baniso -1
Baniso -2
Baniso -3
1-
-0.824 Å2
-0 Å2
-0 Å2
2-
-
0.177 Å2
-0 Å2
3-
-
-
0.647 Å2
Refinement step
Cycle: LAST / Resolution: 1.41→15.696 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
921
0
73
83
1077
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.004
0.013
1027
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.017
953
X-RAY DIFFRACTION
r_angle_refined_deg
1.128
1.644
1371
X-RAY DIFFRACTION
r_angle_other_deg
1.254
1.637
2216
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
4.625
5
113
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
31.036
22.182
55
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
11.743
15
170
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
16.238
15
6
X-RAY DIFFRACTION
r_chiral_restr
0.063
0.2
117
X-RAY DIFFRACTION
r_gen_planes_refined
0.004
0.02
1210
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
220
X-RAY DIFFRACTION
r_nbd_refined
0.195
0.2
211
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.165
0.2
799
X-RAY DIFFRACTION
r_nbtor_refined
0.171
0.2
489
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.081
0.2
375
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.109
0.2
63
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.152
0.2
6
X-RAY DIFFRACTION
r_nbd_other
0.137
0.2
53
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.103
0.2
12
X-RAY DIFFRACTION
r_mcbond_it
1.578
3.487
453
X-RAY DIFFRACTION
r_mcbond_other
1.564
3.471
451
X-RAY DIFFRACTION
r_mcangle_it
2.579
7.807
566
X-RAY DIFFRACTION
r_mcangle_other
2.577
7.824
566
X-RAY DIFFRACTION
r_scbond_it
2.305
4.029
573
X-RAY DIFFRACTION
r_scbond_other
2.303
4.038
574
X-RAY DIFFRACTION
r_scangle_it
3.807
8.68
805
X-RAY DIFFRACTION
r_scangle_other
3.805
8.695
806
X-RAY DIFFRACTION
r_lrange_it
5.622
32.061
1157
X-RAY DIFFRACTION
r_lrange_other
5.579
31.769
1143
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
Fsc free
Fsc work
% reflection obs (%)
WRfactor Rwork
1.41-1.446
0.275
94
0.276
1616
0.276
1712
0.805
0.823
99.8832
0.261
1.446-1.486
0.311
89
0.248
1592
0.251
1683
0.843
0.836
99.8812
0.227
1.486-1.528
0.282
68
0.235
1583
0.237
1659
0.819
0.879
99.5178
0.201
1.528-1.575
0.252
81
0.218
1491
0.219
1581
0.865
0.889
99.4307
0.178
1.575-1.626
0.238
86
0.221
1444
0.222
1537
0.904
0.906
99.5446
0.173
1.626-1.682
0.219
83
0.209
1413
0.21
1506
0.886
0.916
99.336
0.158
1.682-1.745
0.223
71
0.203
1371
0.204
1452
0.921
0.927
99.3113
0.16
1.745-1.815
0.233
54
0.191
1326
0.193
1385
0.927
0.935
99.639
0.149
1.815-1.895
0.227
80
0.181
1263
0.184
1348
0.933
0.945
99.6291
0.151
1.895-1.986
0.16
65
0.17
1204
0.169
1276
0.959
0.958
99.4514
0.149
1.986-2.092
0.159
61
0.159
1156
0.159
1226
0.961
0.965
99.2659
0.148
2.092-2.216
0.183
60
0.15
1090
0.151
1166
0.961
0.968
98.6278
0.146
2.216-2.366
0.179
52
0.156
1046
0.157
1112
0.955
0.965
98.741
0.158
2.366-2.55
0.159
46
0.153
972
0.153
1031
0.968
0.97
98.7391
0.159
2.55-2.787
0.163
49
0.152
899
0.152
960
0.97
0.968
98.75
0.163
2.787-3.103
0.211
49
0.17
810
0.172
875
0.953
0.959
98.1714
0.19
3.103-3.56
0.188
39
0.168
742
0.169
788
0.963
0.964
99.1117
0.196
3.56-4.305
0.194
34
0.176
636
0.177
672
0.972
0.973
99.7024
0.204
4.305-5.872
0.184
24
0.191
524
0.19
550
0.968
0.968
99.6364
0.239
5.872-15.696
0.213
19
0.238
349
0.237
374
0.947
0.957
98.3957
0.269
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj

 Assembly
Assembly

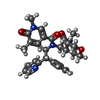
 Mass: 502.605 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C29H34N4O4 / Feature type: SUBJECT OF INVESTIGATION
Mass: 502.605 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C29H34N4O4 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2
 Mass: 354.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34O8 / Comment: precipitant*YM
Mass: 354.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34O8 / Comment: precipitant*YM Mass: 18.015 Da / Num. of mol.: 83 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 83 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID30B / Wavelength: 0.966 Å
/ Beamline: ID30B / Wavelength: 0.966 Å Processing
Processing