 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7e7n: Crystal structure of RSL mutant-R17A/R108A/R199A in complex with R3F -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7e7n | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of RSL mutant-R17A/R108A/R199A in complex with R3F | ||||||
 Components Components | Fucose-binding lectin protein,Fucose-binding lectin protein,Fucose-binding lectin protein | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / Complex / Lectin / Fucose / Rhodamine | ||||||
| Function / homology | Fucose-specific lectin / Fungal fucose-specific lectin / carbohydrate binding / metal ion binding / Chem-R3F / Fucose-binding lectin protein / Fucose-binding lectin protein Function and homology information Function and homology information | ||||||
| Biological species |  Ralstonia solanacearum (bacteria) Ralstonia solanacearum (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å | ||||||
 Authors Authors | Li, L. / Chen, G.S. | ||||||
 Citation Citation | Journal: ACS Nano / Year: 2023 Title: Spatiotemporal Landscape for the Sophisticated Transformation of Protein Assemblies Defined by Multiple Supramolecular Interactions. Authors: Long Li / Zhen Li / Ziying Wang / Shuyu Chen / Rongying Liu / Xuyang Xu / Zhi Zhang / Linfei Ye / Yu Ding / Quan Luo / Sheng Cao / Lei Zhang / Anne Imberty / Guosong Chen /   Abstract: Precise protein assemblies not only constitute a series of living machineries but also provide an advanced class of biomaterials. Previously, we developed the inducing ligand strategy to generate ...Precise protein assemblies not only constitute a series of living machineries but also provide an advanced class of biomaterials. Previously, we developed the inducing ligand strategy to generate various fixed protein assemblies, without the formation of noncovalent interactions between proteins. Here, we demonstrated that controlling the symmetry and number of supramolecular interactions introduced on protein surfaces could direct the formation of unspecific interactions between proteins and induce various nanoscale assemblies, including coiling nanowires, nanotubes, and nanosheets, without manipulation of the protein's native surfaces. More importantly, these nanoscale assemblies could spontaneously evolve into more ordered architectures, crystals. We further showed that the transformation from the introduced supramolecular interactions to the interactions formed between proteins was crucial for pathway selection and outcomes of evolution. These findings reveal a transformation mechanism of protein self-assembly that has not been exploited before and may provide an approach to generate complex and transformable biomacromolecular self-assemblies. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7e7n.cif.gz | 123.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7e7n.ent.gz | 94.7 KB | Display | PDB format |
| PDBx/mmJSON format | 7e7n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e7/7e7nftp://data.pdbj.org/pub/pdb/validation_reports/e7/7e7n | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7e7rC  7e7tC  7e7uC  7e7vC  7e7wC  4csdS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








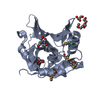


-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29091.568 Da / Num. of mol.: 1 / Mutation: R17A,R108A,R199A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Ralstonia solanacearum (bacteria)Gene: E7Z57_08365, RSP795_21825, RSP822_19650, RUN39_v1_50103 Production host: | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical | 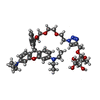  Mass: 803.940 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C43H57N5O10 / Feature type: SUBJECT OF INVESTIGATION Mass: 803.940 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C43H57N5O10 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 150 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 150 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | N | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 49.02 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: small tubes / pH: 7.5 Details: 20 mM Tris-HCl, 100 mM of NaCl pH 7.5, Micro centrifuge tube sequentially put with RSL solution, pure buffer, the ligand solution |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL17U1 / Wavelength: 0.979183 Å | ||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jun 21, 2020 | ||||||||||||||||||||||||||||||
| Radiation | Monochromator: M / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.979183 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.5→39.04 Å / Num. obs: 40510 / % possible obs: 93.8 % / Redundancy: 3.5 % / CC1/2: 0.999 / Rmerge(I) obs: 0.043 / Rpim(I) all: 0.027 / Rrim(I) all: 0.051 / Net I/σ(I): 14.2 / Num. measured all: 143079 / Scaling rejects: 46 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4CSD Resolution: 1.55→39.04 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.93 / SU B: 2.871 / SU ML: 0.051 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.086 / ESU R Free: 0.085 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 128.9 Å2 / Biso mean: 19.97 Å2 / Biso min: 8.43 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.55→39.04 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.55→1.59 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 2.069 Å / Origin y: 8.765 Å / Origin z: 4.946 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
