Dipeptidylpeptidase1 / Cathepsin C / Cathepsin J / Dipeptidyl peptidase I / DPPI / Dipeptidyl transferase
Mass: 13500.163 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSC, CPPI / Cell line (production host): BTI-TN-5B1-4 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
#2: Protein
Dipeptidylpeptidase1 / Cathepsin C / Cathepsin J / Dipeptidyl peptidase I / DPPI / Dipeptidyl transferase
Mass: 18302.613 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSC, CPPI / Cell line (production host): BTI-TN-5B1-4 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
#3: Protein
Dipeptidylpeptidase1 / Cathepsin C / Cathepsin J / Dipeptidyl peptidase I / DPPI / Dipeptidyl transferase
Mass: 7583.444 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSC, CPPI / Cell line (production host): BTI-TN-5B1-4 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
Mass: 18.015 Da / Num. of mol.: 372 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.81 Å3/Da / Density % sol: 56.2 %
Crystal grow
Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 5.6 / Details: 20.0 % (w/v) PEG 3000 0.1 M sodium citrate pH 5.6
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
Diffraction source
Source: ROTATING ANODE / Type: BRUKER X8 PROTEUM / Wavelength: 1.5418 Å
Detector
Type: Bruker Platinum 135 / Detector: CCD / Date: Dec 1, 2010
Radiation
Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 2→27.9 Å / Num. obs: 29396 / % possible obs: 94 % / Redundancy: 11.9 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 29.8
Reflection shell
Resolution: 2→2.1 Å / Rmerge(I) obs: 0.212 / Mean I/σ(I) obs: 6.9 / % possible all: 61.2
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.02382018/15/10
refinement
PROTEUM2
datareduction
SCALA
datascaling
REFMAC
phasing
Refinement
Resolution: 2→27.505 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.94 / SU B: 5.367 / SU ML: 0.081 / Cross valid method: THROUGHOUT / ESU R: 0.153 / ESU R Free: 0.143 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Rfree
0.1983
1429
-
Rwork
0.1519
-
-
all
0.154
-
-
obs
-
28534
93.97 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
Displacement parameters
Biso mean: 17.937 Å2
Baniso -1
Baniso -2
Baniso -3
1-
-0.053 Å2
0 Å2
0 Å2
2-
-
0.146 Å2
0 Å2
3-
-
-
-0.093 Å2
Refinement step
Cycle: LAST / Resolution: 2→27.505 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
2759
0
128
372
3259
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.013
0.013
3008
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.018
2578
X-RAY DIFFRACTION
r_angle_refined_deg
1.838
1.709
4106
X-RAY DIFFRACTION
r_angle_other_deg
1.451
1.631
6008
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
7.914
5
351
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
32.526
22.886
149
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
14.701
15
439
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
23.52
15
11
X-RAY DIFFRACTION
r_chiral_restr
0.086
0.2
391
X-RAY DIFFRACTION
r_gen_planes_refined
0.011
0.02
3339
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
675
X-RAY DIFFRACTION
r_nbd_refined
0.208
0.2
617
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.187
0.2
2547
X-RAY DIFFRACTION
r_nbtor_refined
0.183
0.2
1463
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.086
0.2
1374
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.209
0.2
319
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_other
0.004
0.2
2
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.234
0.2
17
X-RAY DIFFRACTION
r_nbd_other
0.216
0.2
62
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.312
0.2
28
X-RAY DIFFRACTION
r_mcbond_it
1.031
1.071
1398
X-RAY DIFFRACTION
r_mcbond_other
1.031
1.071
1397
X-RAY DIFFRACTION
r_mcangle_it
1.737
1.597
1745
X-RAY DIFFRACTION
r_mcangle_other
1.737
1.597
1746
X-RAY DIFFRACTION
r_scbond_it
1.248
1.332
1609
X-RAY DIFFRACTION
r_scbond_other
1.248
1.334
1610
X-RAY DIFFRACTION
r_scangle_it
2.069
1.963
2356
X-RAY DIFFRACTION
r_scangle_other
2.069
1.965
2357
X-RAY DIFFRACTION
r_lrange_it
5.718
14.955
3617
X-RAY DIFFRACTION
r_lrange_other
5.719
14.969
3618
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Num. reflection all
% reflection obs (%)
WRfactor Rwork
2-2.052
0.228
58
0.179
1398
2209
65.9122
0.145
2.052-2.108
0.206
89
0.181
1614
2146
79.3569
0.145
2.108-2.168
0.231
98
0.166
1837
2099
92.1868
0.136
2.168-2.235
0.224
97
0.154
1870
2036
96.611
0.128
2.235-2.307
0.207
101
0.143
1782
1941
97.0119
0.121
2.307-2.388
0.215
86
0.15
1783
1923
97.1919
0.128
2.388-2.477
0.236
110
0.151
1692
1855
97.1429
0.13
2.477-2.577
0.195
96
0.144
1656
1789
97.9318
0.124
2.577-2.691
0.176
80
0.139
1585
1696
98.1722
0.122
2.691-2.821
0.206
83
0.145
1532
1652
97.7603
0.13
2.821-2.972
0.208
75
0.141
1451
1548
98.5788
0.129
2.972-3.15
0.175
68
0.149
1406
1494
98.6613
0.138
3.15-3.364
0.166
62
0.151
1319
1398
98.784
0.144
3.364-3.629
0.214
68
0.162
1222
1304
98.9264
0.159
3.629-3.969
0.168
64
0.136
1130
1205
99.0871
0.137
3.969-4.426
0.185
47
0.13
1052
1107
99.2773
0.138
4.426-5.089
0.144
49
0.126
919
970
99.7938
0.137
5.089-6.18
0.233
44
0.174
803
853
99.2966
0.182
6.18-8.527
0.26
34
0.189
640
679
99.2636
0.199
8.527-27.546
0.218
20
0.239
408
433
98.8453
0.267
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
1.2538
0.2377
0.4134
1.3245
0.1856
1.3487
-0.0381
0.0016
0.0873
-0.078
0.0004
0.0818
-0.1397
-0.1067
0.0378
0.0563
0.0205
-0.0058
0.0215
0.0144
0.0314
-6.84
-25.289
-20.185
2
0.923
0.0793
0.0138
1.0331
0.192
0.8387
-0.0134
0.0002
-0.0303
-0.0418
0.0057
-0.109
-0.0719
0.1223
0.0077
0.0296
-0.0195
0.0096
0.03
0.0135
0.0373
17.601
-30.779
-17.978
3
0.7666
-0.0855
-0.1195
1.0635
-0.0514
0.768
-0.0114
-0.0939
0.0735
0.0689
0.0054
-0.1148
-0.1698
0.169
0.006
0.0468
-0.0411
-0.0128
0.0572
0.0014
0.028
20.239
-24.406
-8.854
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection
Auth asym-ID
Auth seq-ID
1
X-RAY DIFFRACTION
1
ALL
A
1 - 118
2
X-RAY DIFFRACTION
2
ALL
B
206 - 367
3
X-RAY DIFFRACTION
3
ALL
C
372 - 439
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION / Resolution: 2 Å
X-RAY DIFFRACTION / Resolution: 2 Å  Authors
Authors Poland, 1items
Poland, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj










 Assembly
Assembly

 Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2
Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2
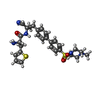

 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 551.723 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H33N5O3S2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 551.723 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H33N5O3S2 / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation Processing
Processing