 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ifb: Crystal structure of polymerase acid protein (PA) from Influenza ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ifb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of polymerase acid protein (PA) from Influenza A virus, WILSON-SMITH/1933 (H1N1) bound to follow on fragment EBSI-4719 5-chloro-2-(1H-imidazol-1-yl)aniline | ||||||
 Components Components | Polymerase acidic protein | ||||||
 Keywords Keywords | VIRAL PROTEIN / NIAID / structural genomics / flu / fragment screening / STD NMR / Seattle Structural Genomics Center for Infectious Disease / SSGCID | ||||||
| Function / homology |  Function and homology information Function and homology informationRNA polymerase activity / negative regulation of viral transcription / negative stranded viral RNA replication / negative regulation of viral genome replication / cap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / protein import into nucleus / endonuclease activity / Hydrolases; Acting on ester bonds / host cell cytoplasm ...RNA polymerase activity / negative regulation of viral transcription / negative stranded viral RNA replication / negative regulation of viral genome replication / cap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / protein import into nucleus / endonuclease activity / Hydrolases; Acting on ester bonds / host cell cytoplasm / symbiont-mediated suppression of host gene expression / viral translational frameshifting / hydrolase activity / host cell nucleus / DNA-templated transcription / RNA binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Influenza A virus Influenza A virus | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.45 Å | ||||||
 Authors Authors | Seattle Structural Genomics Center for Infectious Disease (SSGCID) | ||||||
 Citation Citation | Journal: To Be Published Title: Fragment screening by STD NMR identifies novel site binders against influenza A virus polymerase PA Authors: Pierce, P. / Muruthi, M.M. / Abendroth, J. / Moen, S.O. / Begley, D.W. / Davies, D.R. / Marathias, V.M. / Staker, B.L. / Myler, P.J. / Lorimer, D.D. / Edwards, T.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ifb.cif.gz | 177.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ifb.ent.gz | 137.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5ifb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/if/5ifbftp://data.pdbj.org/pub/pdb/validation_reports/if/5ifb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5ieqC  5if2C  5if5C  5if7C  5if8C  5ifcC  5ifdC  4iujS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 53096.723 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Influenza A virus (strain A/Wilson-Smith/1933 H1N1)Strain: A/Wilson-Smith/1933 H1N1 / Gene: PA / Production host:  |
|---|---|
| #2: Chemical | ChemComp-6AV / 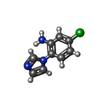  Mass: 193.633 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H8ClN3 Mass: 193.633 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H8ClN3 |
| #3: Chemical | ChemComp-DMS /   Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM |
| #4: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 131 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 131 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 51.94 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 20 mg/mL protein against Morpheus screen condition E10 with 10% PEG 8000, 20% EG, 0.03 M each ethylene glycol, 0.1 M Trizma/bicine pH 8.5, crystal tracking ID 256486e10, unique puck ID lum0-1 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08ID-1 / Wavelength: 0.99985 Å / Beamline: 08ID-1 / Wavelength: 0.99985 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Sep 11, 2014 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.99985 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.45→47.685 Å / Num. obs: 21735 / % possible obs: 99.6 % / Observed criterion σ(I): -3 / Redundancy: 5.2 % / Biso Wilson estimate: 37.26 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.089 / Net I/σ(I): 14.58 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4IUJ Resolution: 2.45→47.685 Å / SU ML: 0.29 / Cross valid method: NONE / σ(F): 1.36 / Phase error: 21.65
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 114.52 Å2 / Biso mean: 45.4789 Å2 / Biso min: 15.91 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.45→47.685 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 8
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
