 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4mk2: 3-(5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile bound to ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4mk2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | 3-(5-hydroxy-6-oxo-1,6-dihydropyridin-3-yl)benzonitrile bound to influenza 2009 pH1N1 endonuclease | ||||||
 Components Components | POLYMERASE PA | ||||||
 Keywords Keywords | RNA BINDING PROTEIN/INHIBITOR / CAP-SNATCHING / RNA BINDING PROTEIN / RNA BINDING PROTEIN-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationcap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / endonuclease activity / Hydrolases; Acting on ester bonds / host cell cytoplasm / symbiont-mediated suppression of host gene expression / viral translational frameshifting / viral RNA genome replication / hydrolase activity / DNA-templated transcription ...cap snatching / symbiont-mediated suppression of host mRNA transcription via inhibition of RNA polymerase II activity / endonuclease activity / Hydrolases; Acting on ester bonds / host cell cytoplasm / symbiont-mediated suppression of host gene expression / viral translational frameshifting / viral RNA genome replication / hydrolase activity / DNA-templated transcription / host cell nucleus / RNA binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   INFLUENZA A VIRUS INFLUENZA A VIRUS | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Bauman, J.D. / Patel, D. / Das, K. / Arnold, E. | ||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2013 Title: Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. Authors: Bauman, J.D. / Patel, D. / Baker, S.F. / Vijayan, R.S. / Xiang, A. / Parhi, A.K. / Martinez-Sobrido, L. / Lavoie, E.J. / Das, K. / Arnold, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4mk2.cif.gz | 139.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4mk2.ent.gz | 111.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4mk2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mk/4mk2ftp://data.pdbj.org/pub/pdb/validation_reports/mk/4mk2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ln7C  4m4qC  4m5oC  4m5qC  4m5rC  4m5vC  4mk1C  4mk5C C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 28505.270 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 1-204 Source method: isolated from a genetically manipulated source Source: (gene. exp.) INFLUENZA A VIRUS (A/LIMA/WRAIR1695P/2009(H1N1))Gene: PA / Plasmid: PCDF-2 / Production host:  |
|---|
-Non-polymers , 5 types, 220 molecules 


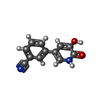

| #2: Chemical |  Mass: 54.938 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mn#3: Chemical | ChemComp-SO4 / |  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-EDO / |  Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#5: Chemical | ChemComp-28B / |  Mass: 212.204 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H8N2O2 Mass: 212.204 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H8N2O2#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 214 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 51.91 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.7 Details: 200 MM MES, 27% PEG8K, 200 MM AMMONIUM SULFATE, 1 MM MANGANESE CHLORIDE, 10 MM MAGNESIUM ACETATE, 10 MM TAURINE, AND 50 MM SODIUM FLUORIDE, pH 6.7, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 0.917 / Beamline: F1 / Wavelength: 0.917 |
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Oct 4, 2012 |
| Radiation | Monochromator: HORIZONTAL BENT SI(111), ASYMMETRICALLY CUT WITH WATER COOLED CU BLOCK Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.917 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→50 Å / Num. obs: 48860 / % possible obs: 99.7 % / Observed criterion σ(I): -3 / Redundancy: 3.5 % / Rmerge(I) obs: 0.048 / Net I/σ(I): 20.61 |
| Reflection shell | Resolution: 1.85→1.88 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.523 / Mean I/σ(I) obs: 1.8 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.85→40.241 Å / SU ML: 0.19 / σ(F): 28.32 / Phase error: 19.23 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→40.241 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
