| 登録情報 | データベース: PDB / ID: 4eox
|
|---|
| タイトル | X-ray Structure of Polypeptide Deformylase Bound to a Acylprolinamide inhibitor |
|---|
 要素 要素 | Peptide deformylase |
|---|
 キーワード キーワード | hydrolase/hydrolase inhibitor / Alpha-beta / peptide deformylase metal ion binding / hydrolase-hydrolase inhibitor complex |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
peptide deformylase / : / peptide deformylase activity / translation / metal ion binding類似検索 - 分子機能 Peptide Deformylase / Peptide deformylase / Peptide deformylase / Peptide deformylase superfamily / Polypeptide deformylase / Alpha-Beta Complex / Alpha Beta類似検索 - ドメイン・相同性 Chem-0S5 / NICKEL (II) ION / Peptide deformylase類似検索 - 構成要素 |
|---|
| 生物種 |   Streptococcus pneumoniae (肺炎レンサ球菌) Streptococcus pneumoniae (肺炎レンサ球菌) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.783 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.783 Å |
|---|
 データ登録者 データ登録者 | Ward, P. / Campobasso, N. |
|---|
 引用 引用 | ジャーナル: Bioorg.Med.Chem.Lett. / 年: 2012
タイトル: Acylprolinamides: a new class of peptide deformylase inhibitors with in vivo antibacterial activity.
著者: Axten, J.M. / Medina, J.R. / Blackledge, C.W. / Duquenne, C. / Grant, S.W. / Bobko, M.A. / Peng, T. / Miller, W.H. / Pinckney, T. / Gallagher, T.F. / Kulkarni, S. / Lewandowski, T. / Van ...著者: Axten, J.M. / Medina, J.R. / Blackledge, C.W. / Duquenne, C. / Grant, S.W. / Bobko, M.A. / Peng, T. / Miller, W.H. / Pinckney, T. / Gallagher, T.F. / Kulkarni, S. / Lewandowski, T. / Van Aller, G.S. / Zonis, R. / Ward, P. / Campobasso, N. |
|---|
| 履歴 | | 登録 | 2012年4月16日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2012年5月30日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2013年1月2日 | Group: Database references |
|---|
| 改定 1.2 | 2024年2月28日 | Group: Data collection / Database references / Derived calculations
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_struct_conn_angle / struct_conn / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj 集合体
集合体

 分子量: 58.693 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ni
分子量: 58.693 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ni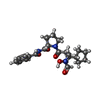
 分子量: 415.483 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C22H29N3O5
分子量: 415.483 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C22H29N3O5 分子量: 18.015 Da / 分子数: 153 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 153 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 21-ID-F / 波長: 0.97872 Å
/ ビームライン: 21-ID-F / 波長: 0.97872 Å 解析
解析