 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4cqf: Crystal structure of Schistosoma mansoni HDAC8 complexed with a m... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4cqf | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Schistosoma mansoni HDAC8 complexed with a mercaptoacetamide inhibitor | ||||||
 Components Components | HISTONE DEACETYLASE 8 | ||||||
 Keywords Keywords | HYDROLASE / STRUCTURAL PROTEIN / EUKARYOTES / PLATYHELMINTHS / EPIGENETICS / HISTONE DEACETYLASES / INHIBITION | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone deacetylase activity, hydrolytic mechanism / histone deacetylase / heterochromatin formation / DNA-templated transcription / metal ion binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Marek, M. / Romier, C. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2014 Title: Molecular Basis for the Anti-Parasitic Activity of a Mercaptoacetamide Derivative that Inhibits Histone Deacetylase 8 (Hdac8) from the Human Pathogen Schistosoma Mansoni Authors: Stolfa, D.A. / Marek, M. / Lancelot, J. / Hauser, A.T. / Walter, A. / Leproult, E. / Melesina, J. / Rumpf, T. / Wurtz, J.M. / Cavarelli, J. / Sippl, W. / Pierce, R.J. / Romier, C. / Jung, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4cqf.cif.gz | 652.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4cqf.ent.gz | 540.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4cqf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cq/4cqfftp://data.pdbj.org/pub/pdb/validation_reports/cq/4cqf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4bz6S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 50444.875 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|
-Non-polymers , 6 types, 485 molecules 

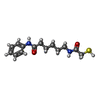
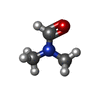


| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: K#4: Chemical | ChemComp-9Z8 /  Mass: 280.386 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C14H20N2O2S Mass: 280.386 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C14H20N2O2S#5: Chemical | ChemComp-DMF /  Mass: 73.094 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C3H7NO Mass: 73.094 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C3H7NO#6: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C3H8O3#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 451 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | THE GSLVPR MOTIF AT THE END OF THE SEQUENCE CORRESPONDS TO A BAMHI CLONING SITE (GS) FOLLOWED BY A ...THE GSLVPR MOTIF AT THE END OF THE SEQUENCE CORRESPOND |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.5 Å3/Da / Density % sol: 19 % / Description: NONE |
|---|---|
| Crystal grow | Details: 0.2 M NA,K L-TARTRATE, 21% (W/V) PEG3350 |
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 |
| Detector | Type: RIGAKU SATURN 944 / Detector: CCD / Date: Sep 20, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→50 Å / Num. obs: 80076 / % possible obs: 96.3 % / Observed criterion σ(I): 2 / Redundancy: 3.8 % / Biso Wilson estimate: 33.74 Å2 / Rmerge(I) obs: 0.11 / Net I/σ(I): 15.2 |
| Reflection shell | Resolution: 2.3→2.34 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.34 / Mean I/σ(I) obs: 4.2 / % possible all: 93.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4BZ6 Resolution: 2.3→32.89 Å / Cor.coef. Fo:Fc: 0.9306 / Cor.coef. Fo:Fc free: 0.8976 / SU R Cruickshank DPI: 0.321 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.3 / SU Rfree Blow DPI: 0.213 / SU Rfree Cruickshank DPI: 0.221 Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. RESIDUE TYPES WITHOUT CCP4 ATOM TYPE IN LIBRARY=ZN K. NUMBER OF ATOMS WITH PROPER CCP4 ATOM TYPE=13636. NUMBER WITH APPROX DEFAULT CCP4 ATOM TYPE=0. ...Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. RESIDUE TYPES WITHOUT CCP4 ATOM TYPE IN LIBRARY=ZN K. NUMBER OF ATOMS WITH PROPER CCP4 ATOM TYPE=13636. NUMBER WITH APPROX DEFAULT CCP4 ATOM TYPE=0. NUMBER TREATED BY BAD NON-BONDED CONTACTS=12.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.65 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.298 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→32.89 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.36 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
