 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4c4g: Structure-based design of orally bioavailable pyrrolopyridine inh... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4c4g | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure-based design of orally bioavailable pyrrolopyridine inhibitors of the mitotic kinase MPS1 | ||||||
 Components Components | DUAL SPECIFICITY PROTEIN KINASE TTK | ||||||
 Keywords Keywords | TRANSFERASE / PROTEIN KINASE / MITOSIS / STRUCTURE-BASED DESIGN | ||||||
| Function / homology |  Function and homology information Function and homology informationprotein localization to meiotic spindle midzone / meiotic spindle assembly checkpoint signaling / repair of mitotic kinetochore microtubule attachment defect / kinetochore binding / female meiosis chromosome segregation / protein localization to kinetochore / dual-specificity kinase / spindle organization / mitotic spindle assembly checkpoint signaling / positive regulation of SMAD protein signal transduction ...protein localization to meiotic spindle midzone / meiotic spindle assembly checkpoint signaling / repair of mitotic kinetochore microtubule attachment defect / kinetochore binding / female meiosis chromosome segregation / protein localization to kinetochore / dual-specificity kinase / spindle organization / mitotic spindle assembly checkpoint signaling / positive regulation of SMAD protein signal transduction / protein serine/threonine/tyrosine kinase activity / mitotic spindle organization / chromosome segregation / kinetochore / spindle / protein tyrosine kinase activity / protein serine kinase activity / protein serine/threonine kinase activity / positive regulation of cell population proliferation / ATP binding / identical protein binding / nucleus / membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å | ||||||
 Authors Authors | Naud, S. / Westwood, I.M. / Faisal, A. / Sheldrake, P. / Bavetsias, V. / Atrash, B. / Liu, M. / Hayes, A. / Schmitt, J. / Wood, A. ...Naud, S. / Westwood, I.M. / Faisal, A. / Sheldrake, P. / Bavetsias, V. / Atrash, B. / Liu, M. / Hayes, A. / Schmitt, J. / Wood, A. / Choi, V. / Boxall, K. / Mak, G. / Gurden, M. / Valenti, M. / de Haven Brandon, A. / Henley, A. / Baker, R. / McAndrew, C. / Matijssen, B. / Burke, R. / Eccles, S.A. / Raynaud, F.I. / Linardopoulos, S. / van Montfort, R. / Blagg, J. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2013 Title: Structure-Based Design of Orally Bioavailable 1H-Pyrrolo[3, 2-C]Pyridine Inhibitors of the Mitotic Kinase Monopolar Spindle 1 (Mps1). Authors: Naud, S. / Westwood, I.M. / Faisal, A. / Sheldrake, P.W. / Bavetsias, V. / Atrash, B. / Cheung, K.J. / Liu, M. / Hayes, A. / Schmitt, J. / Wood, A. / Choi, V. / Boxall, K. / Mak, G. / ...Authors: Naud, S. / Westwood, I.M. / Faisal, A. / Sheldrake, P.W. / Bavetsias, V. / Atrash, B. / Cheung, K.J. / Liu, M. / Hayes, A. / Schmitt, J. / Wood, A. / Choi, V. / Boxall, K. / Mak, G. / Gurden, M. / Valenti, M. / De-Haven-Brandon, A. / Henley, A. / Baker, R. / Mcandrew, C. / Matijssen, B. / Burke, R. / Hoelder, S. / Eccles, S.A. / Raynaud, F.I. / Linardopoulos, S. / Van Montfort, R.L.M. / Blagg, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4c4g.cif.gz | 119.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4c4g.ent.gz | 91.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4c4g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4c4g_validation.pdf.gz | 892.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4c4g_full_validation.pdf.gz | 894.4 KB | Display | |
| Data in XML | 4c4g_validation.xml.gz | 11.8 KB | Display | |
| Data in CIF | 4c4g_validation.cif.gz | 15 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c4/4c4gftp://data.pdbj.org/pub/pdb/validation_reports/c4/4c4g | HTTPS FTP |
-Related structure data
| Related structure data |  4c4eC  4c4fC  4c4hC  4c4iC  4c4jC  4bi1S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36195.234 Da / Num. of mol.: 1 / Fragment: KINASE DOMAIN, RESIDUES 519-808 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-7RO / 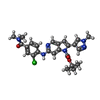  Mass: 494.973 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H27ClN6O3 Mass: 494.973 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H27ClN6O3 | ||||||||
| #3: Chemical |   Mass: 310.384 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H30O7 Mass: 310.384 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H30O7#4: Chemical |   Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | THE SEQUENCE INCLUDING HEXAHISTID | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.8 Å3/Da / Density % sol: 68 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: 30-35% AQUEOUS PEG300 ONLY, pH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9762 / Beamline: I03 / Wavelength: 0.9762 |
| Detector | Type: DECTRIS PILATUS / Detector: PIXEL / Date: Apr 18, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9762 Å / Relative weight: 1 |
| Reflection | Resolution: 2.65→41.04 Å / Num. obs: 13230 / % possible obs: 99.1 % / Observed criterion σ(I): 1.5 / Redundancy: 4.6 % / Biso Wilson estimate: 99.99 Å2 / Rmerge(I) obs: 0.04 / Net I/σ(I): 15.1 |
| Reflection shell | Resolution: 2.65→2.78 Å / Redundancy: 4.3 % / Rmerge(I) obs: 1.15 / Mean I/σ(I) obs: 1.6 / % possible all: 96.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4BI1 Resolution: 2.65→41.04 Å / Cor.coef. Fo:Fc: 0.9494 / Cor.coef. Fo:Fc free: 0.937 / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE LIGAND USED FOR SOAKING WAS 3- CHLORO-N,N-DIMETHYL-4-(2- (1-METHYL-1H-PYRAZOL-4-YL)-1H- PYRROLO(3,2-C)PYRIDIN-6- YLAMINO)BENZAMIDE BUT THE N-BOC PRECURSOR MODELLED HEREIN WAS DETECTED AS ...Details: THE LIGAND USED FOR SOAKING WAS 3- CHLORO-N,N-DIMETHYL-4-(2- (1-METHYL-1H-PYRAZOL-4-YL)-1H- PYRROLO(3,2-C)PYRIDIN-6- YLAMINO)BENZAMIDE BUT THE N-BOC PRECURSOR MODELLED HEREIN WAS DETECTED AS AN IMPURITY AT LESS THAN 2 PERCENT BY LCMS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 103.05 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.514 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.65→41.04 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.65→2.86 Å / Total num. of bins used: 7
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 4.9899 Å / Origin y: 32.4446 Å / Origin z: 129.678 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: CHAIN A |
