 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4a9i: N-TERMINAL BROMODOMAIN OF HUMAN BRD2 WITH 3-methyl-1,2,3,4- tetra... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4a9i | ||||||
|---|---|---|---|---|---|---|---|
| Title | N-TERMINAL BROMODOMAIN OF HUMAN BRD2 WITH 3-methyl-1,2,3,4- tetrahydroquinazolin-2-one | ||||||
 Components Components | BROMODOMAIN CONTAINING 2 | ||||||
 Keywords Keywords | SIGNALING PROTEIN / INHIBITOR / HISTONE / EPIGENETIC READER | ||||||
| Function / homology |  Function and homology information Function and homology informationacetylation-dependent protein binding / chromatin looping / histone H3K14ac reader activity / histone H4K5ac reader activity / histone H4K12ac reader activity / RUNX3 regulates p14-ARF / positive regulation of T-helper 17 cell lineage commitment / neural tube closure / protein localization to chromatin / nucleosome assembly ...acetylation-dependent protein binding / chromatin looping / histone H3K14ac reader activity / histone H4K5ac reader activity / histone H4K12ac reader activity / RUNX3 regulates p14-ARF / positive regulation of T-helper 17 cell lineage commitment / neural tube closure / protein localization to chromatin / nucleosome assembly / histone binding / spermatogenesis / nuclear speck / chromatin remodeling / protein serine/threonine kinase activity / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / nucleoplasm / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.25 Å | ||||||
 Authors Authors | Chung, C.W. / Bamborough, P. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2012 Title: Fragment-Based Discovery of Bromodomain Inhibitors Part 1: Inhibitor Binding Modes and Implications for Lead Discovery. Authors: Chung, C.W. / Dean, A.W. / Woolven, J.M. / Bamborough, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4a9i.cif.gz | 89.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4a9i.ent.gz | 68.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4a9i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a9/4a9iftp://data.pdbj.org/pub/pdb/validation_reports/a9/4a9i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4a9eC  4a9fC  4a9hC  4a9jC  4a9kC  4a9lC C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17908.727 Da / Num. of mol.: 3 / Fragment: N-TERMINAL BROMODOMAIN (BD1), RESIDUES 67-200 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  #2: Chemical | 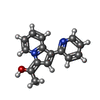  Mass: 237.277 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C15H13N2O Mass: 237.277 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C15H13N2O#3: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-EDO / |   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 38.48 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 293 K / pH: 7 Details: 0.1 M HEPES PH 7.0, 28% PEG 3350, 0.2 M (NH4)2SO4, 20 DEGREES CELSIUS |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Beamline: ID23-1 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Feb 5, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.25→40 Å / Num. obs: 19710 / % possible obs: 97.8 % / Redundancy: 3.4 % / Rmerge(I) obs: 0.1 / Net I/σ(I): 14.6 |
| Reflection shell | Resolution: 2.25→2.29 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.41 / Mean I/σ(I) obs: 2 / % possible all: 96.6 |
- Processing
Processing
| Software | Name: REFMAC / Version: 5.5.0109 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.25→68.04 Å / Cor.coef. Fo:Fc: 0.952 / Cor.coef. Fo:Fc free: 0.914 / SU B: 7.921 / SU ML: 0.191 / Cross valid method: THROUGHOUT / ESU R: 0.313 / ESU R Free: 0.236 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.824 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.25→68.04 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|