 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3j07 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Model of a 24mer alphaB-crystallin multimer | ||||||
 Components Components | Alpha-crystallin B chain | ||||||
 Keywords Keywords | CHAPERONE / sHSP | ||||||
| Function / homology |  Function and homology information Function and homology informationmicrotubule polymerization or depolymerization / negative regulation of intracellular transport / apoptotic process involved in morphogenesis / regulation of programmed cell death / cardiac myofibril / structural constituent of eye lens / tubulin complex assembly / negative regulation of amyloid fibril formation / lens development in camera-type eye / M band ...microtubule polymerization or depolymerization / negative regulation of intracellular transport / apoptotic process involved in morphogenesis / regulation of programmed cell death / cardiac myofibril / structural constituent of eye lens / tubulin complex assembly / negative regulation of amyloid fibril formation / lens development in camera-type eye / M band / muscle organ development / actin filament bundle / negative regulation of reactive oxygen species metabolic process / HSF1-dependent transactivation / stress-activated MAPK cascade / negative regulation of protein-containing complex assembly / muscle contraction / synaptic membrane / response to hydrogen peroxide / glutathione metabolic process / cellular response to gamma radiation / negative regulation of cell growth / Z disc / : / response to estradiol / amyloid-beta binding / response to heat / protein refolding / protein folding / microtubule binding / dendritic spine / perikaryon / response to hypoxia / lysosome / protein stabilization / negative regulation of gene expression / axon / negative regulation of DNA-templated transcription / negative regulation of apoptotic process / protein-containing complex binding / structural molecule activity / cell surface / protein homodimerization activity / protein-containing complex / mitochondrion / extracellular exosome / nucleoplasm / metal ion binding / identical protein binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | SOLID-STATE NMR /  SOLUTION SCATTERING / ELECTRON MICROSCOPY / single particle reconstruction / simulated annealing, molecular dynamics, torsion angle dynamics / negative staining / Resolution: 20 Å SOLUTION SCATTERING / ELECTRON MICROSCOPY / single particle reconstruction / simulated annealing, molecular dynamics, torsion angle dynamics / negative staining / Resolution: 20 Å | ||||||
 Authors Authors | Jehle, S. / Vollmar, B. / Bardiaux, B. / Dove, K.K. / Rajagopal, P. / Gonen, T. / Oschkinat, H. / Klevit, R.E. | ||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2009 Title: The eye lens chaperone alpha-crystallin forms defined globular assemblies. Authors: Jirka Peschek / Nathalie Braun / Titus M Franzmann / Yannis Georgalis / Martin Haslbeck / Sevil Weinkauf / Johannes Buchner /  Abstract: Alpha-crystallins are molecular chaperones that protect vertebrate eye lens proteins from detrimental protein aggregation. alphaB-Crystallin, 1 of the 2 alpha-crystallin isoforms, is also associated ...Alpha-crystallins are molecular chaperones that protect vertebrate eye lens proteins from detrimental protein aggregation. alphaB-Crystallin, 1 of the 2 alpha-crystallin isoforms, is also associated with myopathies and neuropathological diseases. Despite the importance of alpha-crystallins in protein homeostasis, only little is known about their quaternary structures because of their seemingly polydisperse nature. Here, we analyzed the structures of recombinant alpha-crystallins using biophysical methods. In contrast to previous reports, we show that alphaB-crystallin assembles into defined oligomers consisting of 24 subunits. The 3-dimensional (3D) reconstruction of alphaB-crystallin by electron microscopy reveals a sphere-like structure with large openings to the interior of the protein. alphaA-Crystallin forms, in addition to complexes of 24 subunits, also smaller oligomers and large clusters consisting of individual oligomers. This propensity might explain the previously reported polydisperse nature of alpha-crystallin. #1: Journal: Nat Struct Mol Biol / Year: 2010Title: Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Authors: Stefan Jehle / Ponni Rajagopal / Benjamin Bardiaux / Stefan Markovic / Ronald Kühne / Joseph R Stout / Victoria A Higman / Rachel E Klevit / Barth-Jan van Rossum / Hartmut Oschkinat / Abstract: The small heat shock protein alphaB-crystallin (alphaB) contributes to cellular protection against stress. For decades, high-resolution structural studies on oligomeric alphaB have been confounded by ...The small heat shock protein alphaB-crystallin (alphaB) contributes to cellular protection against stress. For decades, high-resolution structural studies on oligomeric alphaB have been confounded by its polydisperse nature. Here, we present a structural basis of oligomer assembly and activation of the chaperone using solid-state NMR and small-angle X-ray scattering (SAXS). The basic building block is a curved dimer, with an angle of approximately 121 degrees between the planes of the beta-sandwich formed by alpha-crystallin domains. The highly conserved IXI motif covers a substrate binding site at pH 7.5. We observe a pH-dependent modulation of the interaction of the IXI motif with beta4 and beta8, consistent with a pH-dependent regulation of the chaperone function. N-terminal region residues Ser59-Trp60-Phe61 are involved in intermolecular interaction with beta3. Intermolecular restraints from NMR and volumetric restraints from SAXS were combined to calculate a model of a 24-subunit alphaB oligomer with tetrahedral symmetry. | ||||||
| History |
| ||||||
| Remark 0 | THIS ENTRY 3J07 CONTAINS A STRUCTURAL MODEL FIT TO AN ELECTRON MICROSCOPY MAP (EMD-1776) DETERMINED ...THIS ENTRY 3J07 CONTAINS A STRUCTURAL MODEL FIT TO AN ELECTRON MICROSCOPY MAP (EMD-1776) DETERMINED ORIGINALLY BY AUTHORS: J.PESCHEK, N.BRAUN, T.M.FRANZMANN, Y.GEORGALIS, M.HASLBECK, S.WEINKAUF, J.BUCHNER |
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3j07.cif.gz | 872.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3j07.ent.gz | 719 KB | Display | PDB format |
| PDBx/mmJSON format | 3j07.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j0/3j07ftp://data.pdbj.org/pub/pdb/validation_reports/j0/3j07 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 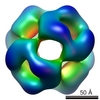 1776M M: map data used to model this data |
|---|---|
| Similar structure data | |
| Other databases |
|






-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: Protein | Mass: 20191.930 Da / Num. of mol.: 24 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CRYAB, CRYA2 / Plasmid: pET16b / Production host:  |
|---|
-Experimental details
-Experiment
| Experiment |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| NMR experiment |
|
- Sample preparation
Sample preparation
| Component | Name: 24mer alphaB-crystallin multimer / Type: COMPLEX / Details: polydisperse oligomer | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer solution | pH: 7.4 / Details: 137mM NaCl, 2.7mM KCl, 12 mM PBS | ||||||||||||||||||||||||||||||||||||
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: YES / Vitrification applied: NO | ||||||||||||||||||||||||||||||||||||
| EM staining | Type: NEGATIVE / Material: Uranyl Acetate | ||||||||||||||||||||||||||||||||||||
| Crystal | Description: HOMOLOGY MODELS AND SPARSE SOLID-STATE NMR DISTANCE RESTRAINTS WERE USED TO DEFINE THE SECONDARY STRUCTURE OF THE N-TERMINAL DOMAIN (UNP RESIDUES 1-65). THE RESTRAINTS ARE GIVEN IN THE ...Description: HOMOLOGY MODELS AND SPARSE SOLID-STATE NMR DISTANCE RESTRAINTS WERE USED TO DEFINE THE SECONDARY STRUCTURE OF THE N-TERMINAL DOMAIN (UNP RESIDUES 1-65). THE RESTRAINTS ARE GIVEN IN THE SUPPORTING INFORMATION OF THE PRIMARY CITATION. A 2D 1H-13C INEPT SPECTRUM WAS RECORDED AND ANALYZED IN ORDER TO IDENTIFY FLEXIBLE RESIDUES IN THE EXTREME N- AND C-TERMINI. SEVERAL MODELS WERE PREPARED WITH DIFFERENT POSITIONS OF FLEXIBLE RESIDUES IN THE INSIDE OF THE SPHERICAL MULTIMER TO FIT THE RADIUS OF GYRATION (RG) MEASURED BY SAXS. THE MODEL THAT BEST FIT THE RG WAS CHOSEN FOR DEPOSITION. | ||||||||||||||||||||||||||||||||||||
| Details |
| ||||||||||||||||||||||||||||||||||||
| Sample |
| ||||||||||||||||||||||||||||||||||||
| Sample conditions | pH: 7.6 / Temperature: 270 K |
-Data collection
| Microscopy | Model: JEOL 100CX / Date: Feb 6, 2008 | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Electron gun | Electron source: OTHER / Accelerating voltage: 100 kV / Illumination mode: FLOOD BEAM | |||||||||||||||||||||||||
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 50000 X / Nominal defocus max: 1200 nm / Nominal defocus min: 900 nm | |||||||||||||||||||||||||
| Image recording | Film or detector model: KODAK SO-163 FILM | |||||||||||||||||||||||||
| NMR spectrometer |
| |||||||||||||||||||||||||
| Soln scatter | Type: x-ray / Buffer name: phosphate, 100 mM NaCl / Conc. range: 0.1-12.6 mg/mL / Data reduction software list: CBF-PRIMUS / Detector type: 2D MAR345 / Mean guiner radius: 5.6 nm / Mean guiner radius esd: 0.2 nm / Num. of time frames: 1 / Protein length: 14.5 / Sample pH: 7.5 / Source beamline: X33 / Source class: Y / Source type: DESY HAMBURG X33 BEAMLINE / Temperature: 293 K |
- Processing
Processing
| Symmetry | Point symmetry: T (tetrahedral) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3D reconstruction | Resolution: 20 Å / Resolution method: FSC 0.5 CUT-OFF / Num. of particles: 2565 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 2KLR Accession code: 2KLR / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
| NMR software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | Method: simulated annealing, molecular dynamics, torsion angle dynamics Software ordinal: 11 Details: THREE DIMERS CONNECTED BY THEIR C-TERMINAL IXI MOTIF WERE FITTED BACK-TO-BACK IN THE EM DENSITY USING CHIMERA. A HEXAMER WAS BUILT FROM MODEL #7 OF PDB ENTRY 2KLR USING NONCRYSTALLOGRAPHIC ...Details: THREE DIMERS CONNECTED BY THEIR C-TERMINAL IXI MOTIF WERE FITTED BACK-TO-BACK IN THE EM DENSITY USING CHIMERA. A HEXAMER WAS BUILT FROM MODEL #7 OF PDB ENTRY 2KLR USING NONCRYSTALLOGRAPHIC SYMMETRY RESTRAINTS GIVEN IN THE PDB FILE. DIMERS WERE CONNECTED BY THEIR C-TERMINAL DOMAIN CONTAINING THE IXI-MOTIF, AND THE LINKER (UNP RESIDUES 151-155) WAS ENERGY MINIMIZED USING XPLOR-NIH. THE BETA1/BETA2(FREE) STRUCTURAL SEGMENT WAS PLACED INTO THE EM DENSITY TO FULFILL THE CROSSLINKS FOR A57, S59, AND T63, SIMILAR TO PDB ENTRY 1VLQ. ALPHA2 HELICES OF TWO MONOMERS WERE DOCKED, DEFINING RESIDUES L32, L33, L37, AND F38 AS INTERACTING RESIDUES USING HADDOCK. THE ALPHA2 HELICAL COMPLEX WAS PLACED IN WEAK EM DENSITY AT THE THREE-FOLD AXIS. THE FLEXIBLE C- AND N-TERMINAL RESIDUES, INCLUDING PARTS OF HELIX ALPHA1, WERE PLACED INSIDE THE 24MER TO FIT THE RADIUS OF GYRATION, MEASURED BY SMALL-ANGLE X-RAY SCATTERING IN AN ITERATIVE PROCESS. ALL FRAGMENTS WERE CONNECTED USING DISCOVERY STUDIO 1.6 (ACCELRYS) AND THE LINKERS WERE ENERGY MINIMIZED. BACK PROJECTIONS WERE CALCULATED FOR MODELS OF ALPHAB MULTIMERS CONTAINING 24, 26, AND 28 SUBUNITS USING THE PROGRAM SPIDER. THE CALCULATED PROJECTIONS ARE SIMILAR TO EACH OTHER, TO THE CLASS SUM IMAGES FROM COLLECTED EM DATA, AND TO THOSE CALCULATED FROM THE PUBLISHED EM DENSITY (EMD-1776). DETAILS ARE GIVEN IN THE PRIMARY CITATION. | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| NMR representative | Selection criteria: lowest energy | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 200 / Conformers submitted total number: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Soln scatter model | Method: FITTING OF MODELS WITH SAXS DATA; Conformer selection criteria: BEST FIT WITH NMR, SAXS, AND EM DATA Details: TO DETERMINE HETEROGENEITY, A PYTHON SCRIPT WAS USED TO MODULATE AND FIT THE THEORETICAL SAXS CURVE OF THE MODEL WITH THE EXPERIMENTAL DATA. THE ALGORITHM THAT WAS APPLIED HAS BEEN PUBLISHED ...Details: TO DETERMINE HETEROGENEITY, A PYTHON SCRIPT WAS USED TO MODULATE AND FIT THE THEORETICAL SAXS CURVE OF THE MODEL WITH THE EXPERIMENTAL DATA. THE ALGORITHM THAT WAS APPLIED HAS BEEN PUBLISHED BY MITTELBACH, R. AND GLATTER, O. (1998) IN J. APPL. CRYSTALLOGR. 31:600-608. FOR DETAILS SEE PRIMARY CITATION. Num. of conformers calculated: 10 / Num. of conformers submitted: 1 / Representative conformer: 1 Software author list: PETOUKHOV, M.V.; SVERGUN, D.I.; SCHWIETERS, C.D. PETTERSON, E.F.; GODDARD, T.D.; ACCELRYS Software list: CHIMERA, PRIMUS, GNOM, XPLOR-NIH, CRYSOL, DISCOVERY STUDIO 1.3; |
