 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3n2p: Crystal structure of human carbonic anhydrase II in complex with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3n2p | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human carbonic anhydrase II in complex with a benzenesulfonamide inhibitor | ||||||
 Components Components | Carbonic anhydrase 2 | ||||||
 Keywords Keywords | LYASE/LYASE INHIBITOR / lyase / zinc metalloenzyme / zinc ligands / benzenesulfonamide / inhibitor. / LYASE-LYASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of dipeptide transmembrane transport / : / regulation of monoatomic anion transport / secretion / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / morphogenesis of an epithelium ...positive regulation of dipeptide transmembrane transport / : / regulation of monoatomic anion transport / secretion / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / morphogenesis of an epithelium / angiotensin-activated signaling pathway / Developmental Lineage of Pancreatic Ductal Cells / carbonic anhydrase / regulation of intracellular pH / carbonate dehydratase activity / positive regulation of synaptic transmission, GABAergic / carbon dioxide transport / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / neuron cellular homeostasis / apical part of cell / myelin sheath / extracellular exosome / zinc ion binding / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.648 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.648 Å | ||||||
 Authors Authors | Avvaru, B.S. / Wagner, J. / Robbins, A.H. / McKenna, R. | ||||||
 Citation Citation | Journal: Chem.Commun.(Camb.) / Year: 2010 Title: Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Authors: Pacchiano, F. / Aggarwal, M. / Avvaru, B.S. / Robbins, A.H. / Scozzafava, A. / McKenna, R. / Supuran, C.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3n2p.cif.gz | 165.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3n2p.ent.gz | 130.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3n2p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n2/3n2pftp://data.pdbj.org/pub/pdb/validation_reports/n2/3n2p | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3mzcC  3n0nC  3n3jC  3n4bC  2iliS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | the biological assembly is a monomer. |
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 29289.062 Da / Num. of mol.: 1 / Fragment: human carbonic anhydrase II / Mutation: wt Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: hCA2 / Production host:  |
|---|
-Non-polymers , 5 types, 222 molecules 



| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
|---|---|
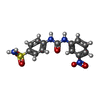
| #3: Chemical | ChemComp-AYX /  Mass: 336.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H12N4O5S Mass: 336.323 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H12N4O5S |
| #4: Chemical | ChemComp-DMS /  Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM |
| #5: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 218 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.96 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.8 Details: 0.05 M tris-Cl, 1.3 M sodium citrate, 5 uL + 5 uL droplet, pH 7.8, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Apr 14, 2010 / Details: mirrors |
| Radiation | Monochromator: Rigaku varimax / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.65→20 Å / Num. all: 28377 / Num. obs: 28377 / % possible obs: 96.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7 % / Biso Wilson estimate: 29.3 Å2 / Rmerge(I) obs: 0.057 / Rsym value: 0.057 / Χ2: 1.034 / Net I/σ(I): 12.8 |
| Reflection shell | Resolution: 1.65→1.71 Å / Redundancy: 6.8 % / Mean I/σ(I) obs: 3.8 / Num. unique all: 2720 / Χ2: 1.053 / % possible all: 93.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2ILI Resolution: 1.648→19.94 Å / Occupancy max: 1 / Occupancy min: 0.48 / SU ML: 0.21 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 40.857 Å2 / ksol: 0.485 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 92.38 Å2 / Biso mean: 22.497 Å2 / Biso min: 9.34 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.648→19.94 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 10
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
