 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3n1w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human FPPS COMPLEX WITH FBS_02 | ||||||
 Components Components | FARNESYL PYROPHOSPHATE SYNTHASE | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / BISPHOSPHONATE / FRAGMENT-BASED SCREENING / TRANSFERASE / ISOPRENE BIOSYNTHESIS / CHOLESTEROL BIOSYNTHESIS / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationLanosterol biosynthesis / zymosterol biosynthetic process / : / : / farnesyl diphosphate biosynthetic process, mevalonate pathway / geranyl diphosphate biosynthetic process / dimethylallyltranstransferase / (2E,6E)-farnesyl diphosphate synthase / geranylgeranyl diphosphate biosynthetic process / trans, trans-farnesyl diphosphate biosynthetic process ...Lanosterol biosynthesis / zymosterol biosynthetic process / : / : / farnesyl diphosphate biosynthetic process, mevalonate pathway / geranyl diphosphate biosynthetic process / dimethylallyltranstransferase / (2E,6E)-farnesyl diphosphate synthase / geranylgeranyl diphosphate biosynthetic process / trans, trans-farnesyl diphosphate biosynthetic process / dimethylallyltranstransferase activity / (2E,6E)-farnesyl diphosphate synthase activity / cholesterol biosynthetic process / Activation of gene expression by SREBF (SREBP) / peroxisome / mitochondrial matrix / RNA binding / metal ion binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.56 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.56 Å | ||||||
 Authors Authors | Rondeau, J.-M. | ||||||
 Citation Citation | Journal: Nat.Chem.Biol. / Year: 2010 Title: Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Authors: Jahnke, W. / Rondeau, J.M. / Cotesta, S. / Marzinzik, A. / Pelle, X. / Geiser, M. / Strauss, A. / Gotte, M. / Bitsch, F. / Hemmig, R. / Henry, C. / Lehmann, S. / Glickman, J.F. / Roddy, T.P. ...Authors: Jahnke, W. / Rondeau, J.M. / Cotesta, S. / Marzinzik, A. / Pelle, X. / Geiser, M. / Strauss, A. / Gotte, M. / Bitsch, F. / Hemmig, R. / Henry, C. / Lehmann, S. / Glickman, J.F. / Roddy, T.P. / Stout, S.J. / Green, J.R. #1: Journal: ChemMedChem / Year: 2006Title: Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs Authors: Rondeau, J.M. / Bitsch, F. / Bourgier, E. / Geiser, M. / Hemmig, R. / Kroemer, M. / Lehmann, S. / Ramage, P. / Rieffel, S. / Strauss, A. / Green, J.R. / Jahnke, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3n1w.cif.gz | 85.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3n1w.ent.gz | 63.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3n1w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n1/3n1wftp://data.pdbj.org/pub/pdb/validation_reports/n1/3n1w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3n1vC  3n3lC  3n45C  3n46C  3n49C  3n5hC  3n5jC  3n6kC C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 40183.855 Da / Num. of mol.: 1 / Fragment: Residues 72-419 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: FDPS, FPS, KIAA1293 / Plasmid: pET28 / Production host:  References: UniProt: P14324, dimethylallyltranstransferase, (2E,6E)-farnesyl diphosphate synthase |
|---|---|
| #2: Chemical | ChemComp-3N2 / (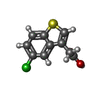  Mass: 226.679 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H7ClO2S Mass: 226.679 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H7ClO2S |
| #3: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.94 Å3/Da / Density % sol: 58.16 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 5.3 Details: 1.2M sodium potassium phosphate, 25% glycerol, pH 5.3, VAPOR DIFFUSION, HANGING DROP, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 0.97933 Å / Beamline: X06SA / Wavelength: 0.97933 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARCCD165 / Detector: CCD / Date: May 7, 2004 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: SAGITALLY FOCUSED SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97933 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.56→40 Å / Num. obs: 15908 / % possible obs: 99.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 7.1 % / Biso Wilson estimate: 54.408 Å2 / Rmerge(I) obs: 0.062 / Net I/σ(I): 18.97 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 2.56→39.36 Å / Rfactor Rfree error: 0.006 / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.763 / Data cutoff high absF: 2787454 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 55.342 Å2 / ksol: 0.37 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 128.87 Å2 / Biso mean: 65.566 Å2 / Biso min: 21.53 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.56→39.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 32
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
