Journal: Mol Cell / Year: 2017 Title: Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Authors: Jan Bednar / Isabel Garcia-Saez / Ramachandran Boopathi / Amber R Cutter / Gabor Papai / Anna Reymer / Sajad H Syed / Imtiaz Nisar Lone / Ognyan Tonchev / Corinne Crucifix / Hervé Menoni / ...Authors: Jan Bednar / Isabel Garcia-Saez / Ramachandran Boopathi / Amber R Cutter / Gabor Papai / Anna Reymer / Sajad H Syed / Imtiaz Nisar Lone / Ognyan Tonchev / Corinne Crucifix / Hervé Menoni / Christophe Papin / Dimitrios A Skoufias / Hitoshi Kurumizaka / Richard Lavery / Ali Hamiche / Jeffrey J Hayes / Patrick Schultz / Dimitar Angelov / Carlo Petosa / Stefan Dimitrov / Abstract: Linker histones associate with nucleosomes to promote the formation of higher-order chromatin structure, but the underlying molecular details are unclear. We investigated the structure of a 197 bp ...Linker histones associate with nucleosomes to promote the formation of higher-order chromatin structure, but the underlying molecular details are unclear. We investigated the structure of a 197 bp nucleosome bearing symmetric 25 bp linker DNA arms in complex with vertebrate linker histone H1. We determined electron cryo-microscopy (cryo-EM) and crystal structures of unbound and H1-bound nucleosomes and validated these structures by site-directed protein cross-linking and hydroxyl radical footprinting experiments. Histone H1 shifts the conformational landscape of the nucleosome by drawing the two linkers together and reducing their flexibility. The H1 C-terminal domain (CTD) localizes primarily to a single linker, while the H1 globular domain contacts the nucleosome dyad and both linkers, associating more closely with the CTD-distal linker. These findings reveal that H1 imparts a strong degree of asymmetry to the nucleosome, which is likely to influence the assembly and architecture of higher-order structures.
History
Deposition
Mar 31, 2017
-
Header (metadata) release
May 17, 2017
-
Map release
May 17, 2017
-
Update
Aug 2, 2017
-
Current status
Aug 2, 2017
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Homo sapiens (human)
Homo sapiens (human) Authors
Authors Citation
Citation


 Structure visualization
Structure visualization Movie viewer
Movie viewer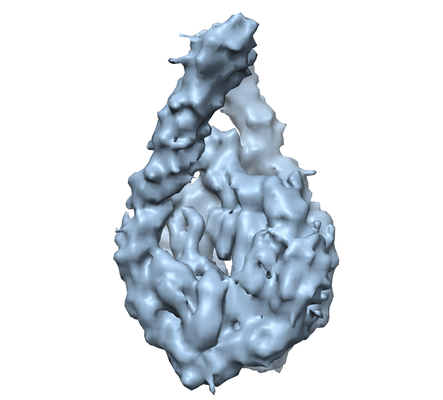
 Downloads & links
Downloads & links emd_3657.png
emd_3657.png http://ftp.pdbj.org/pub/emdb/structures/EMD-3657
http://ftp.pdbj.org/pub/emdb/structures/EMD-3657








 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
