 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2cua | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE CUA DOMAIN OF CYTOCHROME BA3 FROM THERMUS THERMOPHILUS | ||||||
 Components Components | PROTEIN (CUA) | ||||||
 Keywords Keywords | ELECTRON TRANSPORT / CUA CENTER | ||||||
| Function / homology |  Function and homology information Function and homology informationcytochrome-c oxidase / cytochrome-c oxidase activity / respiratory electron transport chain / copper ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |   Thermus thermophilus (bacteria) Thermus thermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.6 Å | ||||||
 Authors Authors | Williams, P.A. / Blackburn, N.J. / Sanders, D. / Bellamy, H. / Stura, E.A. / Fee, J.A. / Mcree, D.E. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1999 Title: The CuA domain of Thermus thermophilus ba3-type cytochrome c oxidase at 1.6 A resolution. Authors: Williams, P.A. / Blackburn, N.J. / Sanders, D. / Bellamy, H. / Stura, E.A. / Fee, J.A. / McRee, D.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2cua.cif.gz | 96.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2cua.ent.gz | 69.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2cua.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cu/2cuaftp://data.pdbj.org/pub/pdb/validation_reports/cu/2cua | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 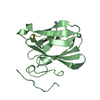
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.0579, -0.444, -0.8942), Vector: |
-Components
| #1: Protein | Mass: 14816.730 Da / Num. of mol.: 2 / Fragment: SOLUBLE CUA-CONTAINING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermus thermophilus (bacteria) / Species (production host): Escherichia coli / Cellular location (production host): PERIPLASM / Production host: #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical |   Mass: 127.092 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cu2 Mass: 127.092 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cu2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 186 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 186 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | THE CUA CENTER COMPRISES TWO CU ATOMS DIRECTLY BONDED, ONE IS FORMALLY 1+ AND THE OTHER 2+ ZINC ...THE CUA CENTER COMPRISES TWO CU ATOMS DIRECTLY BONDED, ONE IS FORMALLY 1+ AND THE OTHER 2+ ZINC AIDED CRYSTALLIS | Sequence details | THE PROTEIN CRYSTALLIS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.33 Å3/Da / Density % sol: 46 % | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: 0.2-20% MPEG 5K 100MM NA CACODYLATE PH 6.5 1MM ZNCL2 | ||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL1-5 / Wavelength: 1.5498,1.3799,1.3780,1.3050,1.0 / Beamline: BL1-5 / Wavelength: 1.5498,1.3799,1.3780,1.3050,1.0 | ||||||||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jan 1, 1998 | ||||||||||||||||||
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 1.6→20 Å / Num. obs: 30110 / % possible obs: 92.4 % / Redundancy: 1.8 % / Rmerge(I) obs: 0.063 / Rsym value: 0.063 / Net I/σ(I): 10.2 | ||||||||||||||||||
| Reflection shell | Resolution: 1.6→1.7 Å / Redundancy: 1.2 % / Rmerge(I) obs: 0.093 / Mean I/σ(I) obs: 8.8 / Rsym value: 0.093 / % possible all: 73.2 | ||||||||||||||||||
| Reflection | *PLUS Num. measured all: 119934 | ||||||||||||||||||
| Reflection shell | *PLUS % possible obs: 73.2 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.6→50 Å / Num. parameters: 8752 / Cross valid method: THROUGHOUT / σ(F): 0 Details: N TERMINUS MAY BE IN A NON-NATIVE CONFORMATION DETERMINED BY THE CRYSTAL PACKING.
| |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→50 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.226 / Rfactor Rfree: 0.296 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
