 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2acz: Complex II (Succinate Dehydrogenase) From E. Coli with Atpenin A5... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2acz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Complex II (Succinate Dehydrogenase) From E. Coli with Atpenin A5 inhibitor co-crystallized at the ubiquinone binding site | ||||||
 Components Components | (Succinate dehydrogenase ...) x 4 | ||||||
 Keywords Keywords | Oxidoreductase/Electron transport / MEMBRANE PROTEIN / Aerobic reparatory Complex II / SQR / succinate:ubiquinone oxidoreductase / AA5 / AT5 / Atpenin A5 / SDH / succinate dehydrogenase / Oxidoreductase-Electron transport COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationsuccinate dehydrogenase activity / respiratory chain complex II (succinate dehydrogenase) / succinate dehydrogenase (quinone) activity / succinate dehydrogenase / cytochrome complex assembly / aerobic electron transport chain / anaerobic respiration / 3 iron, 4 sulfur cluster binding / ubiquinone binding / iron-sulfur cluster binding ...succinate dehydrogenase activity / respiratory chain complex II (succinate dehydrogenase) / succinate dehydrogenase (quinone) activity / succinate dehydrogenase / cytochrome complex assembly / aerobic electron transport chain / anaerobic respiration / 3 iron, 4 sulfur cluster binding / ubiquinone binding / iron-sulfur cluster binding / membrane => GO:0016020 / tricarboxylic acid cycle / aerobic respiration / respiratory electron transport chain / electron transport chain / 2 iron, 2 sulfur cluster binding / flavin adenine dinucleotide binding / 4 iron, 4 sulfur cluster binding / electron transfer activity / heme binding / membrane / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 3.1 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 3.1 Å | ||||||
 Authors Authors | Horsefield, R. / Yankovskaya, V. / Sexton, G. / Whittingham, W. / Shiomi, K. / Omura, S. / Byrne, B. / Cecchini, G. / Iwata, S. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2006 Title: Structural and computational analysis of the quinone-binding site of complex II (succinate-ubiquinone oxidoreductase): a mechanism of electron transfer and proton conduction during ubiquinone reduction. Authors: Horsefield, R. / Yankovskaya, V. / Sexton, G. / Whittingham, W. / Shiomi, K. / Omura, S. / Byrne, B. / Cecchini, G. / Iwata, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2acz.cif.gz | 231.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2acz.ent.gz | 179.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2acz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ac/2aczftp://data.pdbj.org/pub/pdb/validation_reports/ac/2acz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1nekS S: Starting model for refinement |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Succinate dehydrogenase ... , 4 types, 4 molecules ABCD
| #1: Protein | Mass: 64502.766 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P10444, UniProt: P0AC41*PLUS, EC: 1.3.99.1 |
|---|---|
| #2: Protein | Mass: 26800.912 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #3: Protein | Mass: 14313.100 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #4: Protein | Mass: 12874.438 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
-Non-polymers , 8 types, 8 molecules 

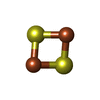



| #5: Chemical | ChemComp-OAA /  Mass: 131.064 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H3O5 Mass: 131.064 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H3O5 |
|---|---|
| #6: Chemical | ChemComp-FAD /  Mass: 785.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM |
| #7: Chemical | ChemComp-FES /  Mass: 175.820 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 175.820 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe2S2 |
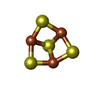
| #8: Chemical | ChemComp-SF4 /  Mass: 351.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 351.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S4 |
| #9: Chemical | ChemComp-F3S /  Mass: 295.795 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe3S4 Mass: 295.795 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe3S4 |
| #10: Chemical | ChemComp-HEB /  Mass: 618.503 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FeN4O4 Mass: 618.503 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FeN4O4 |
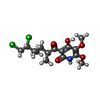
| #11: Chemical | ChemComp-AT5 /  Mass: 366.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H21Cl2NO5 Mass: 366.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H21Cl2NO5 |
| #12: Chemical | ChemComp-CDN /  Mass: 1151.511 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C58H120O17P2 Mass: 1151.511 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C58H120O17P2 |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4 Å3/Da / Density % sol: 68.9 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8.2 Details: Tris-HCl, cacium chloride, Peg 400, pH 8.2, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 170 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.9393 Å / Beamline: ID29 / Wavelength: 0.9393 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: May 10, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9393 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→40 Å / Num. all: 34954 / Num. obs: 34114 / % possible obs: 95.4 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 2 / Redundancy: 4.36 % / Rsym value: 0.078 / Net I/σ(I): 14.7 |
| Reflection shell | Resolution: 3.1→3.16 Å / Mean I/σ(I) obs: 2 / Rsym value: 0.337 / % possible all: 86 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 1NEK Resolution: 3.1→40 Å / Cross valid method: THROUGHOUT / σ(F): -2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 73.7 Å2 | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.1→40 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell | Resolution: 3.1→3.16 Å / Rfactor Rfree: 0.429 / Rfactor Rwork: 0.376 |
