 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-2706: The structure of the immature HIV-1 capsid in intact virus partic... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-2706 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
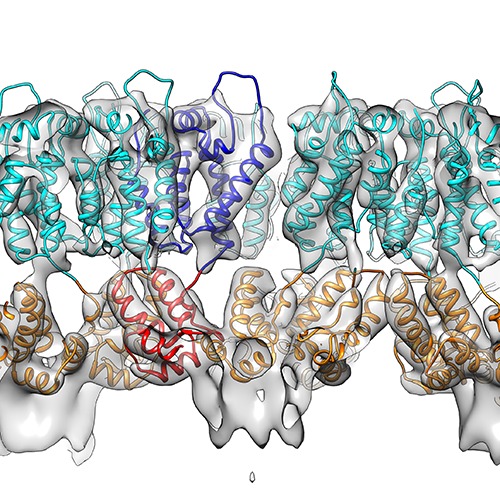
| Title | The structure of the immature HIV-1 capsid in intact virus particles at sub-nm resolution | |||||||||
 Map data Map data | Subtomogram averaging reconstruction of the immature HIV-1 capsid from intact virus particles | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | immature HIV / Retrovirus / Maturation / capsid / Gag | |||||||||
| Function / homology | : / gag protein p24 N-terminal domain / viral process / Retroviral nucleocapsid Gag protein p24, C-terminal domain / Gag protein p24 C-terminal domain / Retrovirus capsid, C-terminal / Retrovirus capsid, N-terminal / viral capsid / p24 Function and homology information Function and homology information | |||||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | |||||||||
| Method | subtomogram averaging / cryo EM / Resolution: 8.8 Å | |||||||||
 Authors Authors | Schur FKM / Hagen WJH / Rumlova M / Ruml T / Mueller B / Kraeusslich H-G / Briggs JAG | |||||||||
 Citation Citation | Journal: Nature / Year: 2015 Title: Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Authors: Florian K M Schur / Wim J H Hagen / Michaela Rumlová / Tomáš Ruml / Barbara Müller / Hans-Georg Kräusslich / John A G Briggs /   Abstract: Human immunodeficiency virus type 1 (HIV-1) assembly proceeds in two stages. First, the 55 kilodalton viral Gag polyprotein assembles into a hexameric protein lattice at the plasma membrane of the ...Human immunodeficiency virus type 1 (HIV-1) assembly proceeds in two stages. First, the 55 kilodalton viral Gag polyprotein assembles into a hexameric protein lattice at the plasma membrane of the infected cell, inducing budding and release of an immature particle. Second, Gag is cleaved by the viral protease, leading to internal rearrangement of the virus into the mature, infectious form. Immature and mature HIV-1 particles are heterogeneous in size and morphology, preventing high-resolution analysis of their protein arrangement in situ by conventional structural biology methods. Here we apply cryo-electron tomography and sub-tomogram averaging methods to resolve the structure of the capsid lattice within intact immature HIV-1 particles at subnanometre resolution, allowing unambiguous positioning of all α-helices. The resulting model reveals tertiary and quaternary structural interactions that mediate HIV-1 assembly. Strikingly, these interactions differ from those predicted by the current model based on in vitro-assembled arrays of Gag-derived proteins from Mason-Pfizer monkey virus. To validate this difference, we solve the structure of the capsid lattice within intact immature Mason-Pfizer monkey virus particles. Comparison with the immature HIV-1 structure reveals that retroviral capsid proteins, while having conserved tertiary structures, adopt different quaternary arrangements during virus assembly. The approach demonstrated here should be applicable to determine structures of other proteins at subnanometre resolution within heterogeneous environments. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_2706.map.gz | 9.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-2706-v30.xmlemd-2706.xml | 11.2 KB 11.2 KB | Display Display | EMDB header |
| Images | emd_2706.tif | 760.9 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2706ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2706 http://ftp.pdbj.org/pub/emdb/structures/EMD-2706ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2706 | HTTPS FTP |
-Related structure data
| Related structure data |  4usnMC  2707C  2708C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_2706.map.gz / Format: CCP4 / Size: 10.2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Subtomogram averaging reconstruction of the immature HIV-1 capsid from intact virus particles | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.025 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : intact HIV-1 particles treated with the protease inhibitor Amprenavir
| Entire | Name: intact HIV-1 particles treated with the protease inhibitor Amprenavir |
|---|---|
| Components |
|
-Supramolecule #1000: intact HIV-1 particles treated with the protease inhibitor Amprenavir
| Supramolecule | Name: intact HIV-1 particles treated with the protease inhibitor Amprenavir type: sample / ID: 1000 / Oligomeric state: Homohexameric / Number unique components: 1 |
|---|
-Supramolecule #1: Human immunodeficiency virus 1
| Supramolecule | Name: Human immunodeficiency virus 1 / type: virus / ID: 1 / Name.synonym: HIV-1 / NCBI-ID: 11676 / Sci species name: Human immunodeficiency virus 1 / Virus type: VIRION / Virus isolate: SPECIES / Virus enveloped: Yes / Virus empty: No / Syn species name: HIV-1 |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) / synonym: VERTEBRATES Homo sapiens (human) / synonym: VERTEBRATES |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | subtomogram averaging |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 6.5 / Details: 25mM MES pH 6.5, 150mM NaCl |
|---|---|
| Grid | Details: C-Flat 2/2-2C glow discharged |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Instrument: FEI VITROBOT MARK II Method: Degassed C-Flat 2/2-2C grids were glow discharged for 30 seconds at 20 mA. Virus solution was diluted in PBS containing 10nm colloidal gold. 2 ul of this mixture was applied to a grid. Blotting time: 2 seconds |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at nominal working magnification |
| Specialist optics | Energy filter - Name: GATAN GIF 2002 |
| Date | Aug 13, 2013 |
| Image recording | Category: CCD / Film or detector model: GATAN MULTISCAN / Average electron dose: 40 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 4.0 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 42000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Tilt series - Axis1 - Min angle: -45 ° / Tilt series - Axis1 - Max angle: 60 ° |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | Subtomogram averaging calculations were performed using the AV3 and TOM packages. Subtomograms were extracted from the surface of the virus. |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C6 (6 fold cyclic) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 8.8 Å / Resolution method: OTHER / Software - Name: TOM, AV3 Details: Reconstruction carried out using subtomogram averaging. Subtomogram averaging was performed using scripts from the TOM (Nickell et al, 2005) and AV3 (Foerster et al, 2005) packages. Number subtomograms used: 32455 |
| CTF correction | Details: Phase flipping of individual tilts |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: 1 |
|---|---|
| Software | Name: Chimera, MDFF |
| Details | Structures for the CA-NTD (PDB 1L6N, chain 1) and CA-CTD (PDB 3DS2, one monomer) were rigid body docked into the EM-density using the "Fit in map" option in chimera. The rigid body fit was further refined using Molecular Dynamics Flexible Fitting. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-4usn: |

-Atomic model buiding 2
| Initial model | PDB ID: |
|---|---|
| Software | Name: Chimera, MDFF |
| Details | Structures for the CA-NTD (PDB 1L6N, chain 1) and CA-CTD (PDB 3DS2, one monomer) were rigid body docked into the EM-density using the "Fit in map" option in chimera. The rigid body fit was further refined using Molecular Dynamics Flexible Fitting. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-4usn: |

