 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-23534 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Designed oligomer D2-1.1B | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
| Biological species | synthetic construct (others) | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 19.0 Å | |||||||||
 Authors Authors | Park YJ / Ivan V / Baker D / Veesler D | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2021 Title: Generation of ordered protein assemblies using rigid three-body fusion. Authors: Ivan Vulovic / Qing Yao / Young-Jun Park / Alexis Courbet / Andrew Norris / Florian Busch / Aniruddha Sahasrabuddhe / Hannes Merten / Danny D Sahtoe / George Ueda / Jorge A Fallas / Sara J ...Authors: Ivan Vulovic / Qing Yao / Young-Jun Park / Alexis Courbet / Andrew Norris / Florian Busch / Aniruddha Sahasrabuddhe / Hannes Merten / Danny D Sahtoe / George Ueda / Jorge A Fallas / Sara J Weaver / Yang Hsia / Robert A Langan / Andreas Plückthun / Vicki H Wysocki / David Veesler / Grant J Jensen / David Baker /  Abstract: Protein nanomaterial design is an emerging discipline with applications in medicine and beyond. A long-standing design approach uses genetic fusion to join protein homo-oligomer subunits via α- ...Protein nanomaterial design is an emerging discipline with applications in medicine and beyond. A long-standing design approach uses genetic fusion to join protein homo-oligomer subunits via α-helical linkers to form more complex symmetric assemblies, but this method is hampered by linker flexibility and a dearth of geometric solutions. Here, we describe a general computational method for rigidly fusing homo-oligomer and spacer building blocks to generate user-defined architectures that generates far more geometric solutions than previous approaches. The fusion junctions are then optimized using Rosetta to minimize flexibility. We apply this method to design and test 92 dihedral symmetric protein assemblies using a set of designed homodimers and repeat protein building blocks. Experimental validation by native mass spectrometry, small-angle X-ray scattering, and negative-stain single-particle electron microscopy confirms the assembly states for 11 designs. Most of these assemblies are constructed from designed ankyrin repeat proteins (DARPins), held in place on one end by α-helical fusion and on the other by a designed homodimer interface, and we explored their use for cryogenic electron microscopy (cryo-EM) structure determination by incorporating DARPin variants selected to bind targets of interest. Although the target resolution was limited by preferred orientation effects and small scaffold size, we found that the dual anchoring strategy reduced the flexibility of the target-DARPIN complex with respect to the overall assembly, suggesting that multipoint anchoring of binding domains could contribute to cryo-EM structure determination of small proteins. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_23534.map.gz | 139.4 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-23534-v30.xmlemd-23534.xml | 16.6 KB 16.6 KB | Display Display | EMDB header |
| Images |  emd_23534.png emd_23534.png | 105.2 KB | ||
| Others | emd_23534_additional_1.map.gzemd_23534_half_map_1.map.gzemd_23534_half_map_2.map.gz | 1.1 MB 1.1 MB 1.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-23534ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23534 http://ftp.pdbj.org/pub/emdb/structures/EMD-23534ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23534 | HTTPS FTP |
-Validation report
| Summary document | emd_23534_validation.pdf.gz | 312.6 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_23534_full_validation.pdf.gz | 312.2 KB | Display | |
| Data in XML | emd_23534_validation.xml.gz | 4.8 KB | Display | |
| Data in CIF | emd_23534_validation.cif.gz | 4.9 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-23534ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-23534 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |














-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_23534.map.gz / Format: CCP4 / Size: 2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 6.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
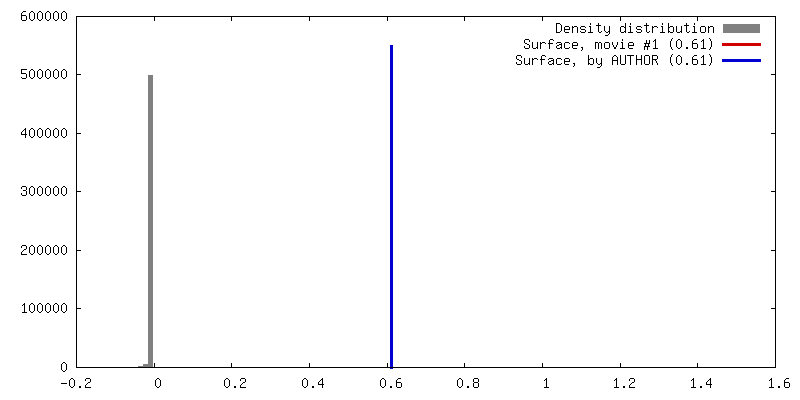
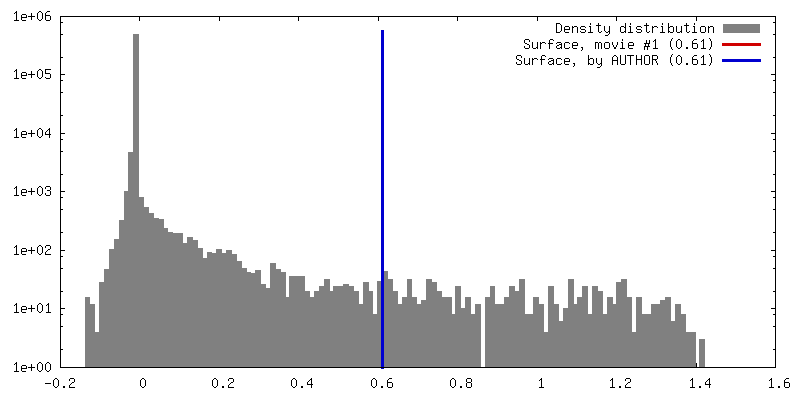
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
-Additional map: #1
| File | emd_23534_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #2
| File | emd_23534_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_23534_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : D2-1.1B
| Entire | Name: D2-1.1B |
|---|---|
| Components |
|
-Supramolecule #1: D2-1.1B
| Supramolecule | Name: D2-1.1B / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Recombinant expression | Organism:  |
-Macromolecule #1: D2-1.1B
| Macromolecule | Name: D2-1.1B / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Sequence | String: MASDYLRLAT EHNKLATEAA SLAAELAASA VTLTVTDPSK TAQEHTELAS RLLEMMSQFL KAAQELTREA IRKEGRNEES EKTLRKSKSS YKALKALLKA IKAIEKGDVE TAVRAAQEAV RLASEAGNNN VLRAVAEVAL AIAKVAEEQG NVEVAVKAAQ VAVSAALNAG ...String: MASDYLRLAT EHNKLATEAA SLAAELAASA VTLTVTDPSK TAQEHTELAS RLLEMMSQFL KAAQELTREA IRKEGRNEES EKTLRKSKSS YKALKALLKA IKAIEKGDVE TAVRAAQEAV RLASEAGNNN VLRAVAEVAL AIAKVAEEQG NVEVAVKAAQ VAVSAALNAG DEDVLKKVAE QASRISKEAE KQGNQEVSKK ALSVSLIAAA ASGDKDLVKD LLESGADVNA SSSDGKTPLH VAAENGHAKV VLLLLEQGAD PNAKDSDGKT PLHLAAENGH AVVVALLLMH GADPNAKDSD GKTPLHLAAE NGHEEVVILL LAMGADPNTS DSDGRTPLDL AREHGNEEVV KVLEDHGGWL EHHHHHH |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 8 |
|---|---|
| Staining | Type: NEGATIVE / Material: UF |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI 12 |
|---|---|
| Image recording | Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 120 kV / Electron source: LAB6 |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
-Image processing
| Startup model | Type of model: OTHER / Details: cryoSPARC ab initio |
|---|---|
| Final reconstruction | Applied symmetry - Point group: D2 (2x2 fold dihedral) / Resolution.type: BY AUTHOR / Resolution: 19.0 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION / Number images used: 84080 |
| Initial angle assignment | Type: PROJECTION MATCHING |
| Final angle assignment | Type: PROJECTION MATCHING |
