 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-20318 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
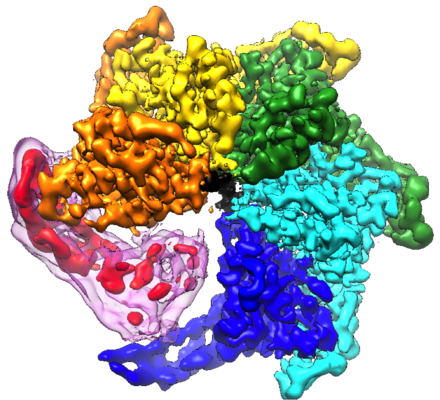
| Title | Msp1-substrate complex in closed conformation | ||||||||||||
 Map data Map data | primary map | ||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | membrane protein / tail-anchored protein / protein quality control / PROTEIN TRANSPORT | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationextraction of mislocalized protein from mitochondrial outer membrane / membrane protein dislocase activity / mitochondrial outer membrane / ATP hydrolysis activity / ATP binding Similarity search - Function | ||||||||||||
| Biological species |  Chaetomium thermophilum (fungus) / Chaetomium thermophilum (fungus) /  | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.1 Å | ||||||||||||
 Authors Authors | Wang L / Myasnikov A | ||||||||||||
| Funding support |  United States, 3 items United States, 3 items
| ||||||||||||
 Citation Citation | Journal: Elife / Year: 2020 Title: Structure of the AAA protein Msp1 reveals mechanism of mislocalized membrane protein extraction. Authors: Lan Wang / Alexander Myasnikov / Xingjie Pan / Peter Walter /  Abstract: The AAA protein Msp1 extracts mislocalized tail-anchored membrane proteins and targets them for degradation, thus maintaining proper cell organization. How Msp1 selects its substrates and firmly ...The AAA protein Msp1 extracts mislocalized tail-anchored membrane proteins and targets them for degradation, thus maintaining proper cell organization. How Msp1 selects its substrates and firmly engages them during the energetically unfavorable extraction process remains a mystery. To address this question, we solved cryo-EM structures of Msp1-substrate complexes at near-atomic resolution. Akin to other AAA proteins, Msp1 forms hexameric spirals that translocate substrates through a central pore. A singular hydrophobic substrate recruitment site is exposed at the spiral's seam, which we propose positions the substrate for entry into the pore. There, a tight web of aromatic amino acids grips the substrate in a sequence-promiscuous, hydrophobic milieu. Elements at the intersubunit interfaces coordinate ATP hydrolysis with the subunits' positions in the spiral. We present a comprehensive model of Msp1's mechanism, which follows general architectural principles established for other AAA proteins yet specializes Msp1 for its unique role in membrane protein extraction. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_20318.map.gz | 2.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-20318-v30.xmlemd-20318.xml | 11.8 KB 11.8 KB | Display Display | EMDB header |
| Images |  emd_20318.png emd_20318.png | 264.2 KB | ||
| Filedesc metadata | emd-20318.cif.gz | 5.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20318ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20318 http://ftp.pdbj.org/pub/emdb/structures/EMD-20318ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20318 | HTTPS FTP |
-Related structure data
| Related structure data |  6pdwMC  6pdyC  6pe0C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_20318.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | primary map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.059 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Msp1-substrate complex in closed conformation
| Entire | Name: Msp1-substrate complex in closed conformation |
|---|---|
| Components |
|
-Supramolecule #1: Msp1-substrate complex in closed conformation
| Supramolecule | Name: Msp1-substrate complex in closed conformation / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 |
|---|---|
| Source (natural) | Organism: Chaetomium thermophilum (fungus) |
| Molecular weight | Theoretical: 240 KDa |
-Macromolecule #1: Membrane-spanning ATPase-like protein
| Macromolecule | Name: Membrane-spanning ATPase-like protein / type: protein_or_peptide / ID: 1 / Number of copies: 5 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Chaetomium thermophilum (fungus) |
| Molecular weight | Theoretical: 42.599152 KDa |
| Recombinant expression | Organism: |
| Sequence | String: GSIAPYLVKI IDPDYEKNER TRIKAQENLR RIRRKQIAEK GDNEDGTDDP SRRRKIDDLV LNEYENQVAL EVVAPEDIPV GFNDIGGLD DIIEELKETI IYPLTMPHLY KHGGALLAAP SGVLLYGPPG CGKTMLAKAV AHESGASFIN LHISTLTEKW Y GDSNKIVR ...String: GSIAPYLVKI IDPDYEKNER TRIKAQENLR RIRRKQIAEK GDNEDGTDDP SRRRKIDDLV LNEYENQVAL EVVAPEDIPV GFNDIGGLD DIIEELKETI IYPLTMPHLY KHGGALLAAP SGVLLYGPPG CGKTMLAKAV AHESGASFIN LHISTLTEKW Y GDSNKIVR AVFSLAKKLQ PSIIFIDEID AVLGTRRSGE HEASGMVKAE FMTLWDGLTS TNASGVPNRI VVLGATNRIN DI DEAILRR MPKQFPVPLP GLEQRRRILE LVLRGTKRDP DFDLDYIARV TAGMSGSDIK ETCRDAAMAP MREYIRQHRA SGK PLSEIN PDDVRGIRTE DFFGRRGGKI LSEIPPRQTG YVVQSKNSSE GGYEEVEDDD EQGTAST UniProtKB: Membrane-spanning ATPase-like protein |
-Macromolecule #2: Unknown peptide
| Macromolecule | Name: Unknown peptide / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 869.063 Da |
| Recombinant expression | Organism: |
| Sequence | String: (UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK) |
-Macromolecule #3: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 3 / Number of copies: 4 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Macromolecule #4: BERYLLIUM TRIFLUORIDE ION
| Macromolecule | Name: BERYLLIUM TRIFLUORIDE ION / type: ligand / ID: 4 / Number of copies: 3 / Formula: BEF |
|---|---|
| Molecular weight | Theoretical: 66.007 Da |
| Chemical component information | 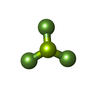 ChemComp-BEF: |
-Macromolecule #5: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 5 / Number of copies: 4 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Details: unspecified |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 70.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: SPOT SCAN / Imaging mode: DIFFRACTION |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: OTHER |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 3.1 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 48861 |
| Initial angle assignment | Type: NOT APPLICABLE |
| Final angle assignment | Type: NOT APPLICABLE |
