 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dyl: 9 ANGSTROM RESOLUTION CRYO-EM RECONSTRUCTION STRUCTURE OF SEMLIKI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dyl | ||||||
|---|---|---|---|---|---|---|---|
| Title | 9 ANGSTROM RESOLUTION CRYO-EM RECONSTRUCTION STRUCTURE OF SEMLIKI FOREST VIRUS (SFV) AND FITTING OF THE CAPSID PROTEIN STRUCTURE IN THE EM DENSITY | ||||||
 Components Components | NUCLEOCAPSID PROTEIN | ||||||
 Keywords Keywords | VIRUS/VIRAL PROTEIN / ALPHAVIRUS / SFV / CRYO-EM / IMAGE RECONSTRUCTION / ENVELOPED VIRUS / CAPSID PROTEIN / ICOSAHEDRAL VIRUS / VIRUS-VIRAL PROTEIN complex | ||||||
| Function / homology |  Function and homology information Function and homology informationtogavirin / T=4 icosahedral viral capsid / virion assembly / small molecule binding / host cell endoplasmic reticulum / channel activity / host cell endosome / monoatomic ion transmembrane transport / clathrin-dependent endocytosis of virus by host cell / symbiont-mediated suppression of host toll-like receptor signaling pathway ...togavirin / T=4 icosahedral viral capsid / virion assembly / small molecule binding / host cell endoplasmic reticulum / channel activity / host cell endosome / monoatomic ion transmembrane transport / clathrin-dependent endocytosis of virus by host cell / symbiont-mediated suppression of host toll-like receptor signaling pathway / host cell Golgi apparatus / entry receptor-mediated virion attachment to host cell / serine-type endopeptidase activity / viral translational frameshifting / fusion of virus membrane with host endosome membrane / viral envelope / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / RNA binding Similarity search - Function | ||||||
| Biological species |   SEMLIKI FOREST VIRUS SEMLIKI FOREST VIRUS | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 9 Å | ||||||
 Authors Authors | Mancini, E.J. / Clarke, M. / Gowen, B.E. / Rutten, T. / Fuller, S.D. | ||||||
 Citation Citation | Journal: Mol Cell / Year: 2000 Title: Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki Forest virus. Authors: E J Mancini / M Clarke / B E Gowen / T Rutten / S D Fuller /  Abstract: Semliki Forest virus serves as a paradigm for membrane fusion and assembly. Our icosahedral reconstruction combined 5276 particle images from 48 cryo-electron micrographs and determined the virion ...Semliki Forest virus serves as a paradigm for membrane fusion and assembly. Our icosahedral reconstruction combined 5276 particle images from 48 cryo-electron micrographs and determined the virion structure to 9 A resolution. The improved resolution of this map reveals an N-terminal arm linking capsid subunits and defines the spike-capsid interaction sites. It illustrates the paired helical nature of the transmembrane segments and the elongated structures connecting them to the spike projecting domains. A 10 A diameter density in the fusion protein lines the cavity at the center of the spike. These clearly visible features combine with the variation in order between the layers to provide a framework for understanding the structural changes during the life cycle of an enveloped virus. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS [A,B,C,D]C IN EACH CHAIN ON SHEET RECORDS ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS [A,B,C,D]C IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dyl.cif.gz | 126.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dyl.ent.gz | 98.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1dyl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dy/1dylftp://data.pdbj.org/pub/pdb/validation_reports/dy/1dyl | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 1015MC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
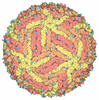

- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 | x 60
|
| 2 |
|
| 3 | x 5
|
| 4 | x 6
|
| 5 | 
|
| Symmetry | Point symmetry: (Schoenflies symbol: I (icosahedral)) |
-Components
| #1: Protein | Mass: 16252.439 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) SEMLIKI FOREST VIRUS / Cell line: BABY HAMSTER KIDNEY 21 / Cellular location: EXTRACELLULAR / References: UniProt: P03315Has protein modification | Y | Sequence details | THE STRUCTURE STUDIED IS FOR THE COMPLETE VIRUS. THE FOLLOWING RESIDUES AND CHAINS ARE NOT REPORTED ...THE STRUCTURE STUDIED IS FOR THE COMPLETE VIRUS. THE FOLLOWING RESIDUES AND CHAINS ARE NOT REPORTED IN THIS ENTRY: MET A 1 TO ASP A 118 SER A 268 TO ARG A 1253 MET B 1 TO ASP B 118 SER B 268 TO ARG B 1253 MET C 1 TO ASP C 118 SER C 268 TO ARG C 1253 MET D 1 TO ASP D 118 SER D 268 TO ARG D 1253 AND CHAINS FOR THE SPIKE GLYCOPROTE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: SEMLIKI FOREST VIRUS / Type: VIRUS |
|---|---|
| Buffer solution | pH: 7.6 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Details: 400 MESH COPPER, HOLEY CARBON, GLOW DISCHARGE, / Grid material: COPPER / Grid mesh size: 400 divisions/in. |
| Vitrification | Instrument: HOMEMADE PLUNGER / Cryogen name: ETHANE Details: PLUNGE VITRIFICATION SAMPLES PREPARED AS THIN LAYERS OF VITREOUS ICE MAINTAINED AT NEAR LIQUID NITROGEN TEMPERATURE IN THE ELECTRON MICROSCOPE WITH A GATAN 626-0300 CRYOTRANSFER HOLDER. |
| Crystal grow | *PLUS Method: other / Details: Fuller, S.D., (1987) Cell. 48, 923. |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: FEI/PHILIPS CM200FEG / Date: Jun 15, 1998 |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: OTHER FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: OTHER |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 50000 X / Nominal defocus max: 7600 nm / Nominal defocus min: 975 nm / Cs: 2 mm |
| Specimen holder | Temperature: 95 K / Tilt angle max: 0 ° / Tilt angle min: 0 ° |
| Image recording | Electron dose: 10 e/Å2 / Film or detector model: KODAK SO-163 FILM |
| Image scans | Num. digital images: 48 |
| Radiation wavelength | Relative weight: 1 |
- Processing
Processing
| EM software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Symmetry | Point symmetry: I (icosahedral) | ||||||||||||
| 3D reconstruction | Method: MODEL-BASED, POLAR-FOURIER- TRANSFORMATION (FULLER ET AL. 1996, J.STRUC.BIOL. 116, 48-55; BAKER AND CHENG 1996, J.STRUC.BIOL. 116, 120- 130) MODEL-BASED CROSS COMMOM LINES SEARCH AND ...Method: MODEL-BASED, POLAR-FOURIER- TRANSFORMATION (FULLER ET AL. 1996, J.STRUC.BIOL. 116, 48-55; BAKER AND CHENG 1996, J.STRUC.BIOL. 116, 120- 130) MODEL-BASED CROSS COMMOM LINES SEARCH AND REFINEMENT (CROWTHER ET AL. 1970, NATURE (LONDON) 226, 421- 425; FULLER ET AL. 1996, J.STRUC.BIOL. 11 48-55;FERLENGHI ET AL. 1998, J.MOL.BIOL. 283, 71-81) Resolution: 9 Å / Num. of particles: 5267 / Nominal pixel size: 2.64 Å / Actual pixel size: 2.52 Å Magnification calibration: THE PIXEL SIZE OF THE CRYO-EM MAP WAS CALIBRATED AGAINST A LOW RESOLUTION DENSITY MAP CALCULATED FROM THE CRYSTAL STRUCTURE OF HRV16. DENSITIES WERE COMPARED BY CROSS- ...Magnification calibration: THE PIXEL SIZE OF THE CRYO-EM MAP WAS CALIBRATED AGAINST A LOW RESOLUTION DENSITY MAP CALCULATED FROM THE CRYSTAL STRUCTURE OF HRV16. DENSITIES WERE COMPARED BY CROSS- CORRELATION WITHIN A SPHERICAL SHELL OF INTERNAL RADIUS 110 ANGSTROMS AND EXTERNAL RADIUS OF 145 ANGSTROMS. Details: THE ORIENTATIONS WERE REFINED BY THE CROSS COMMON LINES LINES METHOD (SIMPLEX) AND THE POLAR FOURIER TRANSFORM METHOD USING A PARALLEL IMPLEMENTATION (PYPFT). THE EFFECTIVE RESOLUTION OF THE ...Details: THE ORIENTATIONS WERE REFINED BY THE CROSS COMMON LINES LINES METHOD (SIMPLEX) AND THE POLAR FOURIER TRANSFORM METHOD USING A PARALLEL IMPLEMENTATION (PYPFT). THE EFFECTIVE RESOLUTION OF THE FINAL RECONSTRUCTED DENSITY WAS DETERMINED TO BE AT LEAST 9 ANGSTROMS, AS MEASURED BY RANDOMLY SPLITTING THE PARTICLES INTO TWO SETS AND CALCULATING THE FOURIER SHELL CORRELATION OBTAINED FROM SEPARATE RECONSTRUCTIONS (HARAUZ AND VAN HEEL 1986, OPTIK 73, 146-156). THE EIGENVALUE SPECTRUM GAVE AN INDICATION OF THE RANDOMNESS OF THE DATA THAT WAS INCLUDED IN THE RECONSTRUCTION. THE COMPLETENESS OF THE DATA WAS VERIFIED IN THAT ALL EIGENVALUES EXCEEDED 100. THE COORDINATES ARE IN THE P, Q, R FRAME IN ANGSTROM UNITS AND CORRESPOND TO ICOSAHEDRAL SYMMETRY AXES. THE ORIGIN IS CHOSEN AT THE CENTER OF THE VIRUS WITH P, Q AND R ALONG MUTUALLY PERPENDICULAR TWO-FOLD AXES OF THE ICOSAHEDRON. THEY SHOULD REMAIN IN THAT FRAME FOR THE EASE OF THE USER IN CREATING THE BIOLOGICALLY SIGNIFICANT VIRAL COMPLEX PARTICLE USING THE 60 ICOSAHEDRAL SYMMETRY OPERATORS. RESIDUES NOT VISIBLE IN THE ORIGINAL CRYSTAL STRUCTURES ARE NOT INCLUDED IN THE CRYO-EM STRUCTURE MODEL. FOR EXAMPLE, C RESIDUES 1-118, ARE NOT VISIBLE IN THE CRYSTAL STRUCTURE (PDB ENTRY 1VCQ) AND THEREFORE ARE NOT INCLUDED IN THE COORDINATES BELOW. Symmetry type: POINT | ||||||||||||
| Atomic model building | Protocol: RIGID BODY FIT / Space: REAL / Target criteria: R-factor Details: METHOD--THE CRYSTAL STRUCTURE OF THE CAPSID PROTEIN FROM CHOI ET AL (1997) PROTEINS 3 27,345-359 (SUBUNIT A OF PDB FILE 1VCQ) WAS PLACED INTO THE CRYO-EM DENSITY MAP. THE CAPSID PROTEIN WAS ...Details: METHOD--THE CRYSTAL STRUCTURE OF THE CAPSID PROTEIN FROM CHOI ET AL (1997) PROTEINS 3 27,345-359 (SUBUNIT A OF PDB FILE 1VCQ) WAS PLACED INTO THE CRYO-EM DENSITY MAP. THE CAPSID PROTEIN WAS FIRST MANUALLY POSITIONED INTO THE CRYO-EM DENSITY CORRESPONDING TO POSITIONS OF THE FOUR INDEPENDENT MONOMER DENSITIES BETWEEN THE INNER LEAFLET OF THE BILAYER AND THE RNA. THESE POSITIONS WERE THEN REFINED BY RIGID BODY REFINEMENT IN REAL SPACE WITH THE PROGRAM EMFIT (CHENG ET AL. 1995, CELL 80, 621-630). THE QUALITY OF THE FIT CAN BE SEEN FROM THE MAP DENSITY WITHIN THE PROTEIN. ALL 4563 ATOMS ARE IN DENSITY OF AT LEAST 4 SIGMA (96.73) ABOVE THE AVERAGE (512.04) , 1167 ATOMS ARE IN DENSITY BETWEEN 4 AND 5 SIGMA, 3174 ATOMS ARE IN DENSITY BETWEEN 5 AND 6 SIGMA, AND 222 ATOMS ARE IN DENSTY OF 6 SIGMA OR ABOVE. THE VARIATION IN DENSITY OVER THE FITTED PROTEIN CAN BE VISUALIZED WITH THE PSEUDO TEMPERATURE FACTOR. THE DENSITY VALUE AT EACH ATOM IS GIVEN IN THE 8TH COLUM (USUALLY THE OCCUPANCY) AS THE NUMBER OF STANDARD DEVIATION ABOVE BACKGROUND. COLUMN NINE (USUALLY THE TEMPERATURE FACTOR) CONTAINS THE VALUE OF THE RELATIVE DENSITY WITHIN THE FITTED PROTEIN SCALED LINEARLY SO THAT THE MINIMUM DENSITY IS 100.0 AND THE MAXIMUM DENSITY IS 1.0. THE ATOMS THAT LIE IN THE LOWER DENSITY REGIONS WILL HAVE THE HIGHEST PSEUDO TEMPERATURE FACTORS. REFINEMENT PROTOCOL--RIGID BODY REFINEMENT | ||||||||||||
| Atomic model building | PDB-ID: 1DYL Accession code: 1DYL / Source name: PDB / Type: experimental model | ||||||||||||
| Refinement | Highest resolution: 9 Å | ||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 9 Å
|
