 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1btw: Episelection: novel KI ~nanomolar inhibitors of serine proteases ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1btw | ||||||
|---|---|---|---|---|---|---|---|
| Title | Episelection: novel KI ~nanomolar inhibitors of serine proteases selected by binding or chemistry on an enzyme surface | ||||||
 Components Components | BETA-TRYPSIN | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / TRIPEPTIDEBORONATE 1 / 3-PROPANEDIOL MONOESTER-INHIBITED / SERINE PROTEINASE / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationtrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / endopeptidase activity / serine-type endopeptidase activity / proteolysis / : / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.7 Å X-RAY DIFFRACTION / Resolution: 1.7 Å | ||||||
 Authors Authors | Stroud, R.M. / Katz, B.A. / Finer-Moore, J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1995 Title: Episelection: novel Ki approximately nanomolar inhibitors of serine proteases selected by binding or chemistry on an enzyme surface. Authors: Katz, B.A. / Finer-Moore, J. / Mortezaei, R. / Rich, D.H. / Stroud, R.M. #1: Journal: Proteins / Year: 1992Title: Solvent Structure in Crystals of Trypsin Determined by X-Ray and Neutron Diffraction Authors: Finer-Moore, J.S. / Kossiakoff, A. / Hurley, J.H. / Earnest, T. / Stroud, R.M. #2: Journal: Proteins / Year: 1991Title: 1.59 Angstrom Structure of Trypsin at 120K: Comparison of Low Temperature and Room Temperature Structures Authors: Earnest, T. / Fauman, E. / Craik, C.S. / Stroud, R.M. #3: Journal: Acta Crystallogr.,Sect.B / Year: 1977Title: Difference Fourier Refinement of the Structure of Dip-Trypsin at 1.5 Angstroms Using a Minicomputer Technique Authors: Chambers, J.L. / Stroud, R.M. #4: Journal: J.Mol.Biol. / Year: 1974Title: Structure and Specific Binding of Trypsin, Comparison of Inhibited Derivatives and a Model for Substrate Binding Authors: Krieger, M. / Kay, L.M. / Stroud, R.M. #5: Journal: Cold Spring Harbor Symp.Quant.Biol. / Year: 1972Title: The Crystal and Molecular Structure of Dip-Inhibited Bovine Trypsin at 2.7 Angstroms Resolution Authors: Stroud, R.M. / Kay, L.M. / Dickerson, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1btw.cif.gz | 77.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1btw.ent.gz | 57.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1btw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bt/1btwftp://data.pdbj.org/pub/pdb/validation_reports/bt/1btw | HTTPS FTP |
|---|
-Related structure data























-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24012.953 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-0ZW / 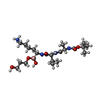  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 475.408 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H44BN4O7 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 475.408 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H44BN4O7References: T-BUTOXY-ALA-VAL-BORO-LYS 1,3-PROPANEDIOL MONOESTER |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 172 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 172 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Nonpolymer details | THE INHIBITOR WAS SYNTHESIZED AS THE PINANEDIOL DIESTER P-TOLUENESULFONATE SALT. THE PROPANEDIOL ...THE INHIBITOR WAS SYNTHESIZE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.85 Å3/Da / Density % sol: 56.86 % |
|---|---|
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: used seeding |
-Data collection
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | Num. obs: 15938 / % possible obs: 50.6 % / Observed criterion σ(I): 1 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 7 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.7→7 Å / σ(F): 3.3 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
