 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1rds: CRYSTAL STRUCTURE OF RIBONUCLEASE MS (AS RIBONUCLEASE T1 HOMOLOGU... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1rds | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF RIBONUCLEASE MS (AS RIBONUCLEASE T1 HOMOLOGUE) COMPLEXED WITH A GUANYLYL-3',5'-CYTIDINE ANALOGUE | ||||||
 Components Components | RIBONUCLEASE MS | ||||||
 Keywords Keywords | HYDROLASE(ENDORIBONUCLEASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationribonuclease T1 / ribonuclease T1 activity / hydrolase activity / RNA binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.8 Å X-RAY DIFFRACTION / Resolution: 1.8 Å | ||||||
 Authors Authors | Nonaka, T. / Nakamura, K.T. / Mitsui, Y. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1993 Title: Crystal structure of ribonuclease Ms (as a ribonuclease T1 homologue) complexed with a guanylyl-3',5'-cytidine analogue. Authors: Nonaka, T. / Nakamura, K.T. / Uesugi, S. / Ikehara, M. / Irie, M. / Mitsui, Y. #1: Journal: FEBS Lett. / Year: 1991Title: Three-Dimensional Structure of Ribonuclease Ms(Asterisk)3'-Guanylic Acid Complex at 2.5 Angstroms Resolution Authors: Nonaka, T. / Mitsui, Y. / Irie, M. / Nakamura, K.T. #2: Journal: Biochemistry / Year: 1990Title: Histidine-40 of Ribonuclease T1 Acts as Base Catalyst When the True Catalytic Base, Glutamic Acid-58,is Replaced by Alanine Authors: Steyaert, J. / Hallenga, K. / Syns, L. / Stanssens, P. #3: Journal: J.Mol.Biol. / Year: 1989Title: Crystallization of a Complex between Ribonuclease Ms and 3'-Guanylic Acid Authors: Nonaka, T. / Mitsui, Y. / Nakamura, K.T. / Watanabe, H. / Ohgi, K. / Irie, M. | ||||||
| History |
| ||||||
| Remark 700 | SHEET ALA 86 (POSITION 1), GLY 87 (POSITION 2) AND ILE 78 (POSITION X) FORM A "CLASSIC TYPE" BETA ...SHEET ALA 86 (POSITION 1), GLY 87 (POSITION 2) AND ILE 78 (POSITION X) FORM A "CLASSIC TYPE" BETA BULGE. ALA 86 AND ILE 78 ALSO OCCUPY POSITIONS 3 AND X+1 OF A "WIDE TYPE" BETA BULGE (SEE REMARK 9). GLU 4 (POSITION 1), TYR 5 (POSITION 2) AND TYR 12 (POSITION X) FORM A "CLASSIC TYPE" BETA BULGE. GLU 84 (POSITION 0), LEU 85 (POSITION 1) ALA 86 (POSITION 3) ILE 78 (POSITION X-1), PHE 79 (POSITION X) AND ASN 80 (POSITION X+1) FORM A "WIDE TYPE" BETA BULGE. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1rds.cif.gz | 35.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1rds.ent.gz | 23.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1rds.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rd/1rdsftp://data.pdbj.org/pub/pdb/validation_reports/rd/1rds | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUE 38 IS A CIS PROLINE. 2: THE PEPTIDE LINKAGE GLY 53 - THR 54 IS IN THE CIS CONFORMATION. |
-Components
| #1: Protein | Mass: 11409.980 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-GPC / 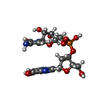  Mass: 590.413 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H24FN8O11P Mass: 590.413 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H24FN8O11P |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 31 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 31 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | SEQUENCE ADVISORY NOTICE: DIFFERENCE BETWEEN SWISS-PROT AND PDB SEQUENCE. SWISS-PROT ENTRY NAME: ...SEQUENCE ADVISORY NOTICE: DIFFERENCE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 42.98 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 6 Å / Num. obs: 7554 / Observed criterion σ(I): 2 / Num. measured all: 23821 / Rmerge(I) obs: 0.0645 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.8→6 Å / Rfactor obs: 0.204 / σ(F): 3 Details: THE FOLLOWING CRITERIA WERE ADOPTED FOR ASSIGNING MAIN-CHAIN HYDROGEN BONDS: THE MAXIMUM DISTANCE BETWEEN AN AMIDE HYDROGEN AND A CARBONYL OXYGEN IS 2.60 ANGSTROMS. THE MAXIMUM DISTANCE ...Details: THE FOLLOWING CRITERIA WERE ADOPTED FOR ASSIGNING MAIN-CHAIN HYDROGEN BONDS: THE MAXIMUM DISTANCE BETWEEN AN AMIDE HYDROGEN AND A CARBONYL OXYGEN IS 2.60 ANGSTROMS. THE MAXIMUM DISTANCE BETWEEN AN AMIDE NITROGEN AND A CARBONYL OXYGEN IS 3.35.ANGSTROMS. THE MINIMUM NHO ANGLE IS 100 DEGREES. THE MINIMUM COH ANGLE IS 100 DEGREES. THE MINIMUM CON ANGLE IS 100 DEGREES. THE ASSIGNMENT OF BETA-TURN TYPES IS BASED ON WILMOT AND THORNTON (C.M.WILMOT AND J.M.THORNTON, PROTEIN ENGINEERING, 3, 479-493, 1990). PHI, PSI ANGLES ARE ALLOWED TO DEVIATE BY 30 DEGREES FROM THEIR IDEAL VALUES. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 6 Å / Num. reflection obs: 6838 / Rfactor obs: 0.204 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
