 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1oxn: Structure and Function Analysis of Peptide Antagonists of Melanom... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1oxn | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure and Function Analysis of Peptide Antagonists of Melanoma Inhibitor of Apoptosis (ML-IAP) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | APOPTOSIS/peptide / zinc binding / peptide complex / apoptosis inhibition / APOPTOSIS-peptide complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of natural killer cell apoptotic process / Regulation of MITF-M-dependent genes involved in apoptosis / lens development in camera-type eye / cysteine-type endopeptidase inhibitor activity / cysteine-type endopeptidase inhibitor activity involved in apoptotic process / negative regulation of tumor necrosis factor-mediated signaling pathway / positive regulation of protein ubiquitination / positive regulation of JNK cascade / RING-type E3 ubiquitin transferase / ubiquitin-protein transferase activity ...regulation of natural killer cell apoptotic process / Regulation of MITF-M-dependent genes involved in apoptosis / lens development in camera-type eye / cysteine-type endopeptidase inhibitor activity / cysteine-type endopeptidase inhibitor activity involved in apoptotic process / negative regulation of tumor necrosis factor-mediated signaling pathway / positive regulation of protein ubiquitination / positive regulation of JNK cascade / RING-type E3 ubiquitin transferase / ubiquitin-protein transferase activity / ubiquitin protein ligase activity / regulation of cell population proliferation / regulation of apoptotic process / regulation of cell cycle / protein ubiquitination / centrosome / negative regulation of apoptotic process / apoptotic process / Golgi apparatus / enzyme binding / nucleoplasm / nucleus / metal ion binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.2 Å | ||||||
 Authors Authors | Franklin, M.C. / Kadkhodayan, S. / Ackerly, H. / Alexandru, D. / Distefano, M.D. / Elliott, L.O. / Flygare, J.A. / Vucic, D. / Deshayes, K. / Fairbrother, W.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Structure and Function Analysis of Peptide Antagonists of Melanoma Inhibitor of Apoptosis (ML-IAP) Authors: Franklin, M.C. / Kadkhodayan, S. / Ackerly, H. / Alexandru, D. / Distefano, M.D. / Elliott, L.O. / Flygare, J.A. / Mausisa, G. / Okawa, D.C. / Ong, D. / Vucic, D. / Deshayes, K. / Fairbrother, W.J. #1: Journal: Curr.Biol. / Year: 2000Title: ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas Authors: Vucic, D. / Stennicke, H.R. / Pisabarro, M.T. / Salvesen, G.S. / Dixit, V.M. #2: Journal: J.Biol.Chem. / Year: 2002Title: SMAC negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ML-IAP) Authors: Vucic, D. / Deshayes, K. / Ackerly, H. / Pisabarro, M.T. / Kadkhodayan, S. / Fairbrother, W.J. / Dixit, V.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1oxn.cif.gz | 125.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1oxn.ent.gz | 96.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1oxn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1oxn_validation.pdf.gz | 704.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1oxn_full_validation.pdf.gz | 711.8 KB | Display | |
| Data in XML | 1oxn_validation.xml.gz | 25.5 KB | Display | |
| Data in CIF | 1oxn_validation.cif.gz | 36.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ox/1oxnftp://data.pdbj.org/pub/pdb/validation_reports/ox/1oxn | HTTPS FTP |
-Related structure data




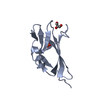







-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 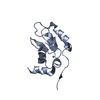
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 
| ||||||||
| 6 | 
| ||||||||
| 7 | 
| ||||||||
| 8 | 
| ||||||||
| 9 | 
| ||||||||
| 10 | 
| ||||||||
| 11 | 
| ||||||||
| 12 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | Each of the five BIR domains in the asymmetric unit represents the biologically active monomer |
-Components
| #1: Protein | Mass: 15749.411 Da / Num. of mol.: 5 / Fragment: BIR domain, residues 63-179 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BIRC7 OR KIAP OR MLIAP OR LIVIN / Plasmid: pET15b / Species (production host): Escherichia coli / Production host:  #2: Protein/peptide | | Mass: 1017.111 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: THE PEPTIDE WAS CHEMICALLY SYNTHESIZED #3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-P33 / |   Mass: 326.383 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H30O8 / Comment: precipitant*YM Mass: 326.383 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H30O8 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 401 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 401 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.99 Å3/Da / Density % sol: 58.5 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 5 Details: sodium acetate, PEG 300, DTT , pH 5.0, VAPOR DIFFUSION, SITTING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.5 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 1.2822, 1.2834, 1.1921 / Beamline: BL9-2 / Wavelength: 1.2822, 1.2834, 1.1921 | ||||||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Apr 12, 2002 / Details: double crystal monochromator | ||||||||||||
| Radiation | Monochromator: double crystal monochromator / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.2→30 Å / Num. all: 74935 / Num. obs: 73906 / % possible obs: 98.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2 % / Biso Wilson estimate: 33.8 Å2 / Rmerge(I) obs: 0.04 / Rsym value: 0.04 / Net I/σ(I): 19.1 | ||||||||||||
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 2 % / Rmerge(I) obs: 0.216 / Mean I/σ(I) obs: 4.4 / Num. unique all: 7395 / Rsym value: 0.216 / % possible all: 99.6 | ||||||||||||
| Reflection | *PLUS Lowest resolution: 20 Å / Rmerge(I) obs: 0.04 | ||||||||||||
| Reflection shell | *PLUS % possible obs: 99.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.2→20 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.927 / SU B: 4.407 / SU ML: 0.114 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.201 / ESU R Free: 0.185 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: The discrepancies between observed reflections and reflections used for refinement is due to merging of Bijvoet mates during refinement. Since the dataset was collected at the zinc anomalous ...Details: The discrepancies between observed reflections and reflections used for refinement is due to merging of Bijvoet mates during refinement. Since the dataset was collected at the zinc anomalous edge, the Bijvoet mates are not identical and represent crystallographically unique reflections.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.119 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.257 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 20 Å / Num. reflection obs: 71265 / % reflection Rfree: 5 % / Rfactor Rfree: 0.225 / Rfactor Rwork: 0.169 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
