 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ojr: L-rhamnulose-1-phosphate aldolase from Escherichia coli (mutant E192A) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ojr | ||||||
|---|---|---|---|---|---|---|---|
| Title | L-rhamnulose-1-phosphate aldolase from Escherichia coli (mutant E192A) | ||||||
 Components Components | RHAMNULOSE-1-PHOSPHATE ALDOLASE | ||||||
 Keywords Keywords | LYASE / ALDOLASE (LYASE) / CLASS II / ZINC ENZYME / C4-TETRAMER / BACTERIAL L-RHAMNOSE METABOLISM / CLEAVAGE OF L-RHAMNULOSE-1- PHOSPHATE TO DIHYDROXYACETONEPHOSPHATE AND L-LACTALDEHYDE | ||||||
| Function / homology |  Function and homology information Function and homology informationrhamnulose-1-phosphate aldolase / rhamnulose-1-phosphate aldolase activity / rhamnose catabolic process / pentose catabolic process / aldehyde-lyase activity / metal ion binding / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å | ||||||
 Authors Authors | Kroemer, M. / Merkel, I. / Schulz, G.E. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Structure and Catalytic Mechanism of L-Rhamnulose-1-Phosphate Aldolase. Authors: Kroemer, M. / Merkel, I. / Schulz, G.E. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2002Title: The Structure of L-Rhamnulose-1-Phosphate Aldolase (Class II) Solved by Low-Resolution Sir Phasing and 20-Fold Ncs Averaging Authors: Kroemer, M. / Schulz, G.E. #2: Journal: J.Mol.Biol. / Year: 2000Title: Structures of L-Fuculose-1-Phosphate Aldolase Mutants Outlining Motions During Catalysis Authors: Joerger, A.C. / Mueller-Dieckmann, C. / Schulz, G.E. #3: Journal: J.Bacteriol. / Year: 1993 Title: Sequencing and Characterization of a Gene Cluster Encoding the Enzymes for L-Rhamnose Metabolism in Escherichia Coli Authors: Moralejo, P. / Egan, S.M. / Hidalgo, E. / Aguilar, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ojr.cif.gz | 152.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ojr.ent.gz | 118.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1ojr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oj/1ojrftp://data.pdbj.org/pub/pdb/validation_reports/oj/1ojr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1gt7S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Sugars , 2 types, 2 molecules A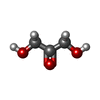
| #1: Protein | Mass: 30116.369 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P32169, rhamnulose-1-phosphate aldolase |
|---|---|
| #4: Sugar | ChemComp-2HA /  Type: saccharide / Mass: 90.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O3 Type: saccharide / Mass: 90.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H6O3 |
-Non-polymers , 5 types, 533 molecules 



| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||||||
|---|---|---|---|---|---|---|---|
| #3: Chemical |  Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4#5: Chemical | ChemComp-DIO / |  Mass: 88.105 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H8O2#6: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 525 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Compound details | ENGINEERED |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.8 Å3/Da / Density % sol: 56 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 4 Details: HANGING DROP WITH 5 MG/ML PROTEIN AND 18% (V/V) DIOXANE. RESERVOIR WITH 35% (V/V) DIOXANE. HAMPTON CRYSTAL SCREEN-2 NO.4, pH 4.00 | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 3 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 0.8468 / Beamline: BW7B / Wavelength: 0.8468 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 15, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8468 Å / Relative weight: 1 |
| Reflection | Resolution: 1.35→30.5 Å / Num. obs: 71387 / % possible obs: 96 % / Redundancy: 3.7 % / Biso Wilson estimate: 10.6 Å2 / Rmerge(I) obs: 0.052 / Net I/σ(I): 8.7 |
| Reflection shell | Resolution: 1.35→1.4 Å / Redundancy: 3.4 % / Rmerge(I) obs: 0.38 / Mean I/σ(I) obs: 1.7 / % possible all: 95 |
| Reflection | *PLUS Highest resolution: 1.35 Å / Lowest resolution: 30.5 Å / % possible obs: 96 % / Redundancy: 3.7 % |
| Reflection shell | *PLUS % possible obs: 95 % / Redundancy: 3.4 % / Num. unique obs: 7660 / Rmerge(I) obs: 0.38 / Mean I/σ(I) obs: 1.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GT7 Resolution: 1.35→30.43 Å / Cor.coef. Fo:Fc: 0.984 / Cor.coef. Fo:Fc free: 0.974 / SU B: 1.647 / SU ML: 0.034 / Cross valid method: THROUGHOUT / ESU R: 0.039 / ESU R Free: 0.04 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. TWO ADDITIONAL DENSITY REGIONS IN THE ACTIVE CENTER WERE FITTED BY A 1:1 MIXTURE OF DIHYDROXYACETONE AND PHOSPHATE AND A 1:1 MIXTURE OF ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. TWO ADDITIONAL DENSITY REGIONS IN THE ACTIVE CENTER WERE FITTED BY A 1:1 MIXTURE OF DIHYDROXYACETONE AND PHOSPHATE AND A 1:1 MIXTURE OF PHOSPHATE AND FIVE WATER MOLECULES, RESPECTIVELY. DIHYDROXYACETONE WAS AN 0.2% (V/V) IMPURITY IN THE AUTOCLAVED CRYO PROTECTANT GLYCEROL. THE PHOSPHATE WAS FROM THE LAST PURIFICATION BUFFER AND HAD ESCAPED DIALYSIS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.43 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.35→30.43 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|