 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hk8: STRUCTURAL BASIS FOR ALLOSTERIC SUBSTRATE SPECIFICITY REGULATION ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hk8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL BASIS FOR ALLOSTERIC SUBSTRATE SPECIFICITY REGULATION IN CLASS III RIBONUCLEOTIDE REDUCTASES: NRDD IN COMPLEX WITH DGTP | |||||||||
 Components Components | ANAEROBIC RIBONUCLEOTIDE-TRIPHOSPHATE REDUCTASE | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / REDUCTASE / ALLOSTERIC REGULATION / SUBSTRATE SPECIFICITY | |||||||||
| Function / homology |  Function and homology information Function and homology informationribonucleoside-triphosphate reductase (formate) / ribonucleoside-triphosphate reductase (thioredoxin) activity / 2'-deoxyribonucleotide biosynthetic process / ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor / DNA replication / metal ion binding Similarity search - Function | |||||||||
| Biological species |  BACTERIOPHAGE T4 (virus) BACTERIOPHAGE T4 (virus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.45 Å | |||||||||
 Authors Authors | Larsson, K.-M. / Andersson, J. / Sjoeberg, B.-M. / Nordlund, P. / Logan, D.T. | |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2003 Title: A Metal-Binding Site in the Catalytic Subunit of Anaerobic Ribonucleotide Reductase. Authors: Logan, D.T. / Mulliez, E. / Larsson, K.-M. / Bodevin, S. / Atta, M. / Garnaud, P.E. / Sjoberg, B.-M. / Fontecave, M. #1: Journal: Structure / Year: 2001Title: Structural basis for allosteric substrate specificity regulation in anaerobic ribonucleotide reductases. Authors: Larsson, K.M. / Andersson, J. / Sjoberg, B.M. / Nordlund, P. / Logan, D.T. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hk8.cif.gz | 132.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hk8.ent.gz | 100.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1hk8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hk/1hk8ftp://data.pdbj.org/pub/pdb/validation_reports/hk/1hk8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b8b S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 68055.594 Da / Num. of mol.: 1 / Fragment: ACTIVE SITE SUBUNIT, RESIDUES 1-605 / Mutation: YES Source method: isolated from a genetically manipulated source Details: SEQUENCE DETERMINATION\: P.YOUNG, M.OHMAN, M.Q.XU. / Source: (gene. exp.) BACTERIOPHAGE T4 (virus) / Plasmid: PET29T4NRDD(G580A) / Production host:  References: UniProt: P07071, ribonucleoside-triphosphate reductase (thioredoxin) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | 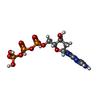  Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3#3: Chemical | ChemComp-ZN / |   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-MN / |   Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 228 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 228 / Source method: isolated from a natural source / Formula: H2OCompound details | CATALYTIC ACTIVITY INVOLVES 2'-DEOXYRIBONUCLEOSIDE TRIPHOSPHATE + OXIDIZED THIOREDOXIN + H(2)O = ...CATALYTIC ACTIVITY INVOLVES 2'-DEOXYRIBON | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.5 Å3/Da / Density % sol: 72.4 % |
|---|---|
| Crystal grow | pH: 7.5 / Details: 30% PEG 400, 02M MGCL2, 0.1M HEPES PH 7.5, 7MM DTT |
| Crystal grow | *PLUS Method: unknown / Details: Logan, D.T., (1999) Science, 283, 1499. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I711 / Wavelength: 1.043 / Beamline: I711 / Wavelength: 1.043 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: May 15, 1999 / Details: BENT MIRROR |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.043 Å / Relative weight: 1 |
| Reflection | Resolution: 2.43→37.8 Å / Num. obs: 44361 / % possible obs: 95.6 % / Observed criterion σ(I): 0 / Redundancy: 3.7 % / Rmerge(I) obs: 0.096 / Net I/σ(I): 15.3 |
| Reflection shell | Resolution: 2.43→2.49 Å / Rmerge(I) obs: 0.274 / Mean I/σ(I) obs: 3.8 / % possible all: 94.1 |
| Reflection | *PLUS Highest resolution: 2.45 Å / Lowest resolution: 20 Å / % possible obs: 95 % / Rmerge(I) obs: 0.076 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1B8B 1b8b Resolution: 2.45→20.08 Å / Cor.coef. Fo:Fc: 0.934 / Cor.coef. Fo:Fc free: 0.901 / SU B: 6.546 / SU ML: 0.148 / Cross valid method: THROUGHOUT / ESU R: 0.244 / ESU R Free: 0.213 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.49 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.45→20.08 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|