 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gqw | ||||||
|---|---|---|---|---|---|---|---|
| Title | Taurine/alpha-ketoglutarate Dioxygenase from Escherichia coli | ||||||
 Components Components | ALPHA-KETOGLUTARATE-DEPENDENT TAURINE DIOXYGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / TAURINE / SULPHUR METABOLISM / OXYGENASE / ALPHA-KETOGLUTARATE / TAUD / TFDA / DIOXYGENASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtaurine catabolic process / taurine dioxygenase complex / taurine dioxygenase / taurine dioxygenase activity / sulfur compound metabolic process / L-ascorbic acid binding / ferrous iron binding / protein homotetramerization / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MIR / Resolution: 3 Å X-RAY DIFFRACTION / MIR / Resolution: 3 Å | ||||||
 Authors Authors | Elkins, J.M. / Ryle, M.J. / Clifton, I.J. / Dunning-Hotopp, J.C. / Lloyd, J.S. / Burzlaff, N.I. / Baldwin, J.E. / Hausinger, R.P. / Roach, P.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: X-Ray Crystal Structure of Escherichia Coli Taurine/Alpha-Ketoglutarate Dioxygenase Complexed to Ferrous Iron and Substrates Authors: Elkins, J.M. / Ryle, M.J. / Clifton, I.J. / Dunning Hotopp, J. / Lloyd, J.S. / Burzlaff, N.I. / Baldwin, J.E. / Hausinger, R.P. / Roach, P.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gqw.cif.gz | 110.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gqw.ent.gz | 88.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1gqw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1gqw_validation.pdf.gz | 465.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1gqw_full_validation.pdf.gz | 488.3 KB | Display | |
| Data in XML | 1gqw_validation.xml.gz | 24.9 KB | Display | |
| Data in CIF | 1gqw_validation.cif.gz | 32.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gq/1gqwftp://data.pdbj.org/pub/pdb/validation_reports/gq/1gqw | HTTPS FTP |
-Related structure data









-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.85901, 0.51171, -0.01591), Vector: |
-Components
| #1: Protein | Mass: 32453.467 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical |   Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe#3: Chemical | 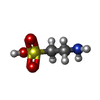  Mass: 125.147 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H7NO3S Mass: 125.147 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H7NO3S#4: Chemical | 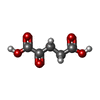  Mass: 146.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 146.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H6O5Compound details | CONVERTS ALPHA KETOGLUTAR | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 60 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 20-28% PEG1000, 20% ETHYLENE GLYCOL, 75MM IMIDAZOLE PH7.5, PROTEIN SOLUTION LOADED WITH FE(II), ALPHA-KETOGLUTARATE, TAURINE, DITHIOTHREITOL, pH 7.50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 17 ℃ / pH: 7 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Dec 15, 2000 / Details: OSMIC |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 3→40.49 Å / Num. obs: 16990 / % possible obs: 99.3 % / Redundancy: 5.9 % / Biso Wilson estimate: 60.4 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 10.5 |
| Reflection shell | Resolution: 3→3.16 Å / Redundancy: 6 % / Rmerge(I) obs: 0.27 / Mean I/σ(I) obs: 2.8 / % possible all: 99.8 |
| Reflection | *PLUS Num. obs: 16727 / Num. measured all: 98529 |
| Reflection shell | *PLUS % possible obs: 99.8 % / Rmerge(I) obs: 0.27 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 3→40.44 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 2733619.8 / Isotropic thermal model: GROUP / Cross valid method: THROUGHOUT / σ(F): 0 Details: RESIDUES WITH DISORDERED SIDE-CHAINS WERE MODELED AS ALANINE
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / ksol: 0.318286 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 63.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→40.44 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Rms dev position: 0.03 Å / Weight position: 300 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3→3.19 Å / Rfactor Rfree error: 0.026 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.32 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.354 |
