 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1fbl: STRUCTURE OF FULL-LENGTH PORCINE SYNOVIAL COLLAGENASE (MMP1) REVE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fbl | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF FULL-LENGTH PORCINE SYNOVIAL COLLAGENASE (MMP1) REVEALS A C-TERMINAL DOMAIN CONTAINING A CALCIUM-LINKED, FOUR-BLADED BETA-PROPELLER | ||||||
 Components Components | FIBROBLAST (INTERSTITIAL) COLLAGENASE (MMP-1) | ||||||
 Keywords Keywords | METALLOPROTEASE | ||||||
| Function / homology |  Function and homology information Function and homology informationCollagen degradation / Activation of Matrix Metalloproteinases / Degradation of the extracellular matrix / interstitial collagenase / Basigin interactions / cellular response to UV-A / collagen catabolic process / extracellular matrix organization / extracellular matrix / positive regulation of protein-containing complex assembly ...Collagen degradation / Activation of Matrix Metalloproteinases / Degradation of the extracellular matrix / interstitial collagenase / Basigin interactions / cellular response to UV-A / collagen catabolic process / extracellular matrix organization / extracellular matrix / positive regulation of protein-containing complex assembly / metalloendopeptidase activity / peptidase activity / proteolysis / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.5 Å | ||||||
 Authors Authors | Li, J. / Brick, P. / Blow, D.M. | ||||||
 Citation Citation | Journal: Structure / Year: 1995 Title: Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four-bladed beta-propeller. Authors: Li, J. / Brick, P. / O'Hare, M.C. / Skarzynski, T. / Lloyd, L.F. / Curry, V.A. / Clark, I.M. / Bigg, H.F. / Hazleman, B.L. / Cawston, T.E. / Blow, D.M. #1: Journal: J.Mol.Biol. / Year: 1989Title: Crystallization and Preliminary X-Ray Analysis of Porcine Synovial Collagenase Authors: Lloyd, L.F. / Skarzynski, T. / Wonacott, A.J. / Cawston, T.E. / Clark, I.M. / Mannix, C.J. / Harper, G.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fbl.cif.gz | 97.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fbl.ent.gz | 72.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1fbl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1fbl_validation.pdf.gz | 745.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1fbl_full_validation.pdf.gz | 754.9 KB | Display | |
| Data in XML | 1fbl_validation.xml.gz | 20.7 KB | Display | |
| Data in CIF | 1fbl_validation.cif.gz | 30 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fb/1fblftp://data.pdbj.org/pub/pdb/validation_reports/fb/1fbl | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42688.355 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#3: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-HTA / | 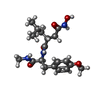  Mass: 379.451 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H29N3O5 Mass: 379.451 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H29N3O5#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 295 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 295 / Source method: isolated from a natural source / Formula: H2OCompound details | THE ENZYME CONSISTS OF AN N-TERMINAL CATALYTIC DOMAIN JOINED BY A LINKING PEPTIDE TO A C-TERMINAL ...THE ENZYME CONSISTS OF AN N-TERMINAL CATALYTIC DOMAIN JOINED BY A LINKING PEPTIDE TO A C-TERMINAL DOMAIN. THE MODEL FOR THE N-TERMINAL DOMAIN (RESIDUES 100 - 260) INCLUDES TWO ZINC IONS AND 3 CALCIUM IONS. ZINC 998 IS AT THE ACTIVE SITE AND COORDINATE | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.97 Å3/Da / Density % sol: 69.01 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal | *PLUS Density % sol: 70.5 % | ||||||||||||||||||
| Crystal grow | *PLUS pH: 7.3 / Method: vapor diffusion, hanging drop | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.876 / Beamline: PX9.6 / Wavelength: 0.876 |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Oct 11, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.876 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→42.2 Å / Num. obs: 22212 / % possible obs: 94.2 % / Redundancy: 3.27 % / Rmerge(I) obs: 0.086 |
| Reflection | *PLUS Num. measured all: 72655 / Rmerge(I) obs: 0.086 |
| Reflection shell | *PLUS % possible obs: 66.7 % / Rmerge(I) obs: 0.23 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.5→9 Å / σ(F): 0 Details: THE CONFORMATION OF THE MAIN CHAIN IS NOT WELL DEFINED FOR RESIDUES 263 - 268, 306 - 311 AND 402 - 409.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→9 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 2.61 Å / Rfactor obs: 0.366 |
