| 登録情報 | データベース: PDB / ID: 1dqa
|
|---|
| タイトル | COMPLEX OF THE CATALYTIC PORTION OF HUMAN HMG-COA REDUCTASE WITH HMG, COA, AND NADP+ |
|---|
 要素 要素 | PROTEIN (HMG-COA REDUCTASE) |
|---|
 キーワード キーワード | OXIDOREDUCTASE / CHOLESTEROL BIOSYNTHESIS / HMG-COA / NADPH |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
hydroxymethylglutaryl-CoA reductase (NADPH) / hydroxymethylglutaryl-CoA reductase (NADPH) activity / sterol biosynthetic process / isopentenyl diphosphate biosynthetic process, mevalonate pathway / GTPase regulator activity / negative regulation of amyloid-beta clearance / coenzyme A binding / farnesyl diphosphate biosynthetic process, mevalonate pathway / zymosterol biosynthetic process / : ...hydroxymethylglutaryl-CoA reductase (NADPH) / hydroxymethylglutaryl-CoA reductase (NADPH) activity / sterol biosynthetic process / isopentenyl diphosphate biosynthetic process, mevalonate pathway / GTPase regulator activity / negative regulation of amyloid-beta clearance / coenzyme A binding / farnesyl diphosphate biosynthetic process, mevalonate pathway / zymosterol biosynthetic process / : / : / Lanosterol biosynthesis / geranylgeranyl diphosphate biosynthetic process / coenzyme A metabolic process / EGR2 and SOX10-mediated initiation of Schwann cell myelination / isoprenoid biosynthetic process / cholesterol biosynthetic process / peroxisomal membrane / cytoplasmic side of endoplasmic reticulum membrane / negative regulation of protein secretion / NADPH binding / Activation of gene expression by SREBF (SREBP) / negative regulation of protein catabolic process / PPARA activates gene expression / endoplasmic reticulum membrane / endoplasmic reticulum類似検索 - 分子機能 HMGR, N-terminal domain / HMGR, N-terminal domain / Hydroxymethylglutaryl-CoA reductase, metazoan / Hydroxymethylglutaryl-CoA reductase, eukaryotic/archaeal type / Hydroxymethylglutaryl-CoA reductase, N-terminal / 3-hydroxy-3-methylglutaryl-coenzyme A Reductase; Chain A, domain 2 / 3-hydroxy-3-methylglutaryl-coenzyme A Reductase; Chain A, domain 2 / Hydroxymethylglutaryl-CoA reductase, class I/II, NAD/NADP-binding domain / Hydroxymethylglutaryl-coenzyme A reductases signature 2. / Hydroxymethylglutaryl-coenzyme A reductases signature 1. ...HMGR, N-terminal domain / HMGR, N-terminal domain / Hydroxymethylglutaryl-CoA reductase, metazoan / Hydroxymethylglutaryl-CoA reductase, eukaryotic/archaeal type / Hydroxymethylglutaryl-CoA reductase, N-terminal / 3-hydroxy-3-methylglutaryl-coenzyme A Reductase; Chain A, domain 2 / 3-hydroxy-3-methylglutaryl-coenzyme A Reductase; Chain A, domain 2 / Hydroxymethylglutaryl-CoA reductase, class I/II, NAD/NADP-binding domain / Hydroxymethylglutaryl-coenzyme A reductases signature 2. / Hydroxymethylglutaryl-coenzyme A reductases signature 1. / Hydroxymethylglutaryl-coenzyme A reductases signature 3. / Hydroxymethylglutaryl-CoA reductase, class I/II / Hydroxymethylglutaryl-CoA reductase, class I/II, NAD/NADP-binding domain superfamily / Hydroxymethylglutaryl-CoA reductase, class I/II, substrate-binding domain superfamily / Hydroxymethylglutaryl-CoA reductase, class I/II, catalytic domain superfamily / Hydroxymethylglutaryl-CoA reductase, class I/II, conserved site / Hydroxymethylglutaryl-coenzyme A reductase / Hydroxymethylglutaryl-coenzyme A reductases family profile. / : / Sterol-sensing domain of SREBP cleavage-activation / Sterol-sensing domain (SSD) profile. / Sterol-sensing domain / Alpha-Beta Plaits / Alpha-Beta Complex / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta類似検索 - ドメイン・相同性 COENZYME A / 3-HYDROXY-3-METHYL-GLUTARIC ACID / NADP NICOTINAMIDE-ADENINE-DINUCLEOTIDE PHOSPHATE / 3-hydroxy-3-methylglutaryl-coenzyme A reductase類似検索 - 構成要素 |
|---|
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 2 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 2 Å |
|---|
 データ登録者 データ登録者 | Istvan, E.S. / Palnitkar, M. / Buchanan, S.K. / Deisenhofer, J. |
|---|
 引用 引用 | ジャーナル: EMBO J. / 年: 2000
タイトル: Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis.
著者: Istvan, E.S. / Palnitkar, M. / Buchanan, S.K. / Deisenhofer, J. |
|---|
| 履歴 | | 登録 | 1999年12月30日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2000年3月8日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2008年4月27日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Version format compliance |
|---|
| 改定 1.3 | 2018年3月14日 | Group: Database references / カテゴリ: struct_ref_seq_dif / Item: _struct_ref_seq_dif.details |
|---|
| 改定 1.4 | 2024年2月7日 | Group: Data collection / Database references / Derived calculations
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj






 集合体
集合体

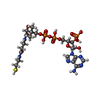
 分子量: 767.534 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C21H36N7O16P3S
分子量: 767.534 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C21H36N7O16P3S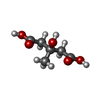
 分子量: 162.141 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C6H10O5
分子量: 162.141 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C6H10O5
 分子量: 743.405 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C21H28N7O17P3
分子量: 743.405 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C21H28N7O17P3 分子量: 18.015 Da / 分子数: 471 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 471 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: BL7-1 / 波長: 1.083
/ ビームライン: BL7-1 / 波長: 1.083  解析
解析