 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bx4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF HUMAN ADENOSINE KINASE AT 1.50 ANGSTROMS | ||||||
 Components Components | PROTEIN (ADENOSINE KINASE) | ||||||
 Keywords Keywords | TRANSFERASE / HUMAN ADENOSINE KINASE | ||||||
| Function / homology |  Function and homology information Function and homology informationdATP biosynthetic process / adenosine kinase / adenosine kinase activity / dAMP salvage / ribonucleoside monophosphate biosynthetic process / deoxyadenosine kinase activity / Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor / Ribavirin ADME / Purine salvage / GMP salvage ...dATP biosynthetic process / adenosine kinase / adenosine kinase activity / dAMP salvage / ribonucleoside monophosphate biosynthetic process / deoxyadenosine kinase activity / Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor / Ribavirin ADME / Purine salvage / GMP salvage / AMP salvage / purine ribonucleoside salvage / purine nucleobase metabolic process / RNA binding / nucleoplasm / ATP binding / metal ion binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.5 Å | ||||||
 Authors Authors | Mathews, I.I. / Erion, M.D. / Ealick, S.E. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Structure of human adenosine kinase at 1.5 A resolution. Authors: Mathews, I.I. / Erion, M.D. / Ealick, S.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bx4.cif.gz | 88.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bx4.ent.gz | 66.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1bx4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bx/1bx4ftp://data.pdbj.org/pub/pdb/validation_reports/bx/1bx4 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 38755.219 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human)Description: NATIVE PROTEIN WAS EXPRESSED IN BL21[DE3] CELLS SELENO METHIONINE PROTEIN IN B834[DE3] CELLS Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#3: Chemical |   Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg#4: Chemical | 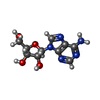  Mass: 267.241 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 267.241 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13N5O4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 354 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 354 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 47 % / Description: MLPHARE WAS USED FOR THE REFINEMENT | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 / Details: 20%PEG 4K, 0.16M MGCL2, 0.1M TRIS, PH 7.5 | |||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 0.929 / Beamline: F1 / Wavelength: 0.929 |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Aug 15, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.929 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→20 Å / Num. obs: 57595 / % possible obs: 98.3 % / Redundancy: 3.7 % / Rmerge(I) obs: 0.048 / Rsym value: 0.048 / Net I/σ(I): 6.3 |
| Reflection shell | Resolution: 1.5→1.58 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.185 / Mean I/σ(I) obs: 3.7 / Rsym value: 0.185 / % possible all: 91.4 |
| Reflection | *PLUS Num. measured all: 259153 |
| Reflection shell | *PLUS % possible obs: 91.4 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.5→20 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / σ(F): 2 Details: TOPOLOGY AND PARAMETER FILES FOR ADENOSINE WERE GENERATED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.57 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARHCSDX.PRO / Topol file: TOPHCSDX.PRO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.8 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 2 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 16.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.267 / % reflection Rfree: 5 % / Rfactor Rwork: 0.247 |
