 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
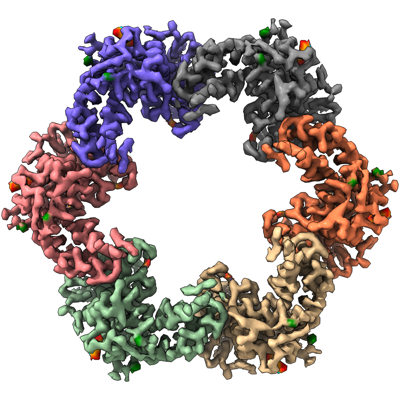
| Title | In situ structure of the Caulobacter crescentus S-layer | |||||||||
 Map data Map data | PostProcessed map with B-factor sharpening | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | RsaA S-layer sub-tomogram averaging Caulobacter / STRUCTURAL PROTEIN | |||||||||
| Function / homology | RsaA N-terminal domain / RTX calcium-binding nonapeptide repeat / RTX calcium-binding nonapeptide repeat (4 copies) / Serralysin-like metalloprotease, C-terminal / calcium ion binding / S-layer protein rsaA Function and homology information Function and homology information | |||||||||
| Biological species |  Caulobacter vibrioides NA1000 (bacteria) Caulobacter vibrioides NA1000 (bacteria) | |||||||||
| Method | subtomogram averaging / cryo EM / Resolution: 3.5 Å | |||||||||
 Authors Authors | von Kuegelgen A / Bharat T | |||||||||
| Funding support |  United Kingdom, 2 items United Kingdom, 2 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2022 Title: A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Authors: Jasenko Zivanov / Joaquín Otón / Zunlong Ke / Andriko von Kügelgen / Euan Pyle / Kun Qu / Dustin Morado / Daniel Castaño-Díez / Giulia Zanetti / Tanmay A M Bharat / John A G Briggs / Sjors H W Scheres /    Abstract: We present a new approach for macromolecular structure determination from multiple particles in electron cryo-tomography (cryo-ET) data sets. Whereas existing subtomogram averaging approaches are ...We present a new approach for macromolecular structure determination from multiple particles in electron cryo-tomography (cryo-ET) data sets. Whereas existing subtomogram averaging approaches are based on 3D data models, we propose to optimise a regularised likelihood target that approximates a function of the 2D experimental images. In addition, analogous to Bayesian polishing and contrast transfer function (CTF) refinement in single-particle analysis, we describe the approaches that exploit the increased signal-to-noise ratio in the averaged structure to optimise tilt-series alignments, beam-induced motions of the particles throughout the tilt-series acquisition, defoci of the individual particles, as well as higher-order optical aberrations of the microscope. Implementation of our approaches in the open-source software package RELION aims to facilitate their general use, particularly for those researchers who are already familiar with its single-particle analysis tools. We illustrate for three applications that our approaches allow structure determination from cryo-ET data to resolutions sufficient for de novo atomic modelling. #1: Journal: BioRxiv / Year: 2022Title: A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0 Authors: Zivanov J / Oton J / Ke Z / von Kuegelgen A / Pyle E / Qu K / Morado D / Castano-Diez D / Zanetti G / Bharat TAM / Briggs JAG / Scheres SHW | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_16183.map.gz | 13.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-16183-v30.xmlemd-16183.xml | 21.8 KB 21.8 KB | Display Display | EMDB header |
| Images |  emd_16183.png emd_16183.png | 175.8 KB | ||
| Masks | emd_16183_msk_1.map | 15.6 MB | Mask map | |
| Filedesc metadata | emd-16183.cif.gz | 7.1 KB | ||
| Others | emd_16183_half_map_1.map.gzemd_16183_half_map_2.map.gz | 7.1 MB 7.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-16183ftp://ftp.pdbj.org/pub/emdb/structures/EMD-16183 http://ftp.pdbj.org/pub/emdb/structures/EMD-16183ftp://ftp.pdbj.org/pub/emdb/structures/EMD-16183 | HTTPS FTP |
-Validation report
| Summary document | emd_16183_validation.pdf.gz | 957.6 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_16183_full_validation.pdf.gz | 957.2 KB | Display | |
| Data in XML | emd_16183_validation.xml.gz | 9.7 KB | Display | |
| Data in CIF | emd_16183_validation.cif.gz | 11.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-16183ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-16183 | HTTPS FTP |
-Related structure data
| Related structure data |  8bqeMC  8bshC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_16183.map.gz / Format: CCP4 / Size: 15.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | PostProcessed map with B-factor sharpening | ||||||||||||||||||||||||||||||||||||
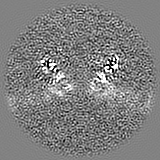
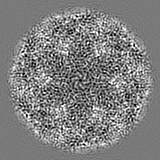
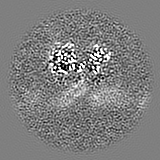
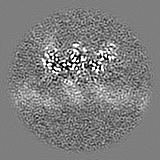
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.35 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)












-Supplemental data
-Mask #1
| File | emd_16183_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: Half map 1
| File | emd_16183_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
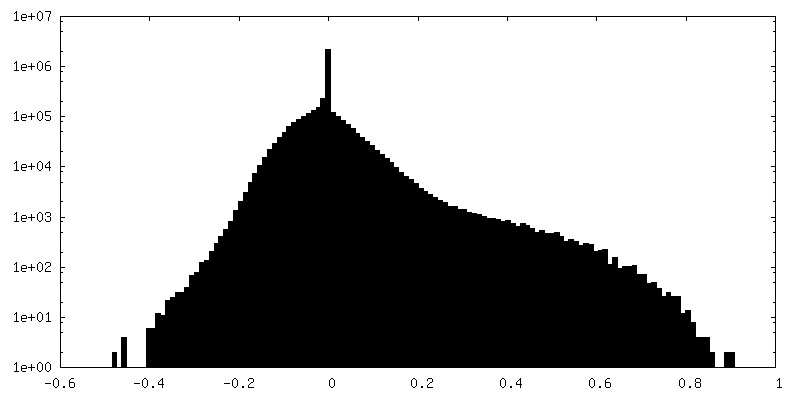
| Annotation | Half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



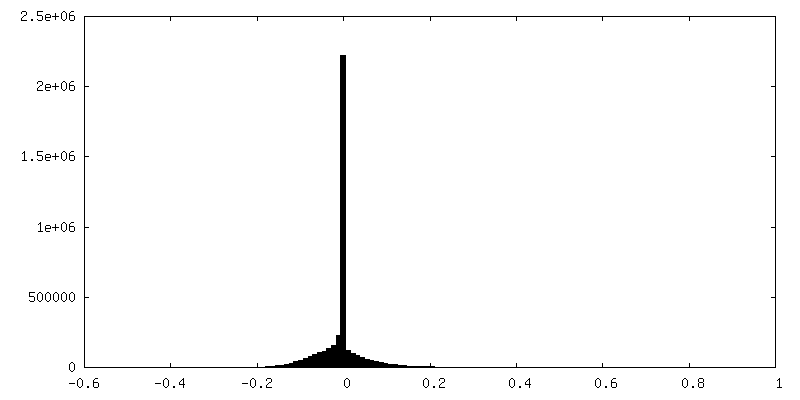
-Half map: Half map 2
| File | emd_16183_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
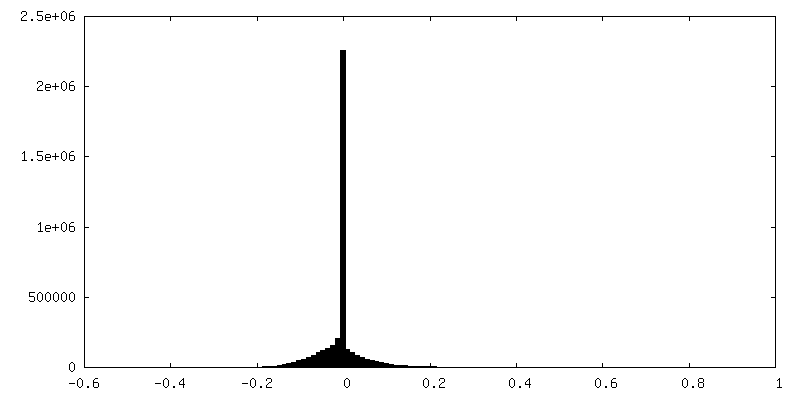
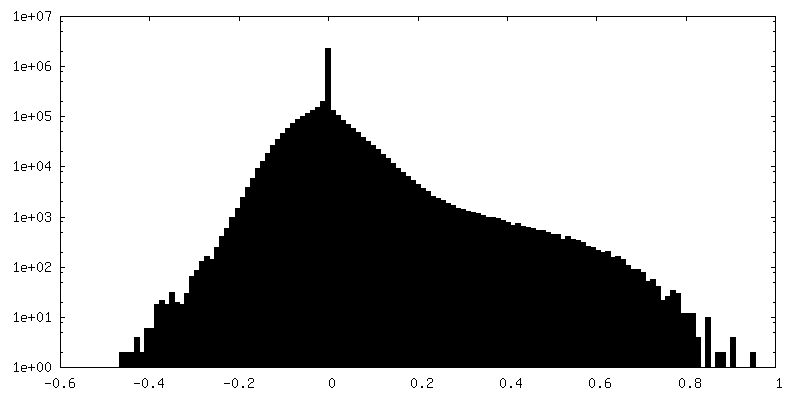
| Annotation | Half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





- Sample components
Sample components
-Entire : Caulobacter crescentus S-layer
| Entire | Name: Caulobacter crescentus S-layer |
|---|---|
| Components |
|
-Supramolecule #1: Caulobacter crescentus S-layer
| Supramolecule | Name: Caulobacter crescentus S-layer / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: #1 / Details: Caulobacter crescentus S-layer |
|---|---|
| Source (natural) | Organism: Caulobacter vibrioides NA1000 (bacteria) / Strain: YB2811 / Location in cell: extra-cellular |
-Macromolecule #1: S-layer protein rsaA
| Macromolecule | Name: S-layer protein rsaA / type: protein_or_peptide / ID: 1 / Details: In-situ S-layer / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Caulobacter vibrioides NA1000 (bacteria) / Strain: NA1000 / CB15N |
| Molecular weight | Theoretical: 98.153906 KDa |
| Recombinant expression | Organism: Caulobacter vibrioides NA1000 (bacteria) |
| Sequence | String: MAYTTAQLVT AYTNANLGKA PDAATTLTLD AYATQTQTGG LSDAAALTNT LKLVNSTTAV AIQTYQFFTG VAPSAAGLDF LVDSTTNTN DLNDAYYSKF AQENRFINFS INLATGAGAG ATAFAAAYTG VSYAQTVATA YDKIIGNAVA TAAGVDVAAA V AFLSRQAN ...String: MAYTTAQLVT AYTNANLGKA PDAATTLTLD AYATQTQTGG LSDAAALTNT LKLVNSTTAV AIQTYQFFTG VAPSAAGLDF LVDSTTNTN DLNDAYYSKF AQENRFINFS INLATGAGAG ATAFAAAYTG VSYAQTVATA YDKIIGNAVA TAAGVDVAAA V AFLSRQAN IDYLTAFVRA NTPFTAAADI DLAVKAALIG TILNAATVSG IGGYATATAA MINDLSDGAL STDNAAGVNL FT AYPSSGV SGSTLSLTTG TDTLTGTANN DTFVAGEVAG AATLTVGDTL SGGAGTDVLN WVQAAAVTAL PTGVTISGIE TMN VTSGAA ITLNTSSGVT GLTALNTNTS GAAQTVTAGA GQNLTATTAA QAANNVAVDG GANVTVASTG VTSGTTTVGA NSAA SGTVS VSVANSSTTT TGAIAVTGGT AVTVAQTAGN AVNTTLTQAD VTVTGNSSTT AVTVTQTAAA TAGATVAGRV NGAVT ITDS AAASATTAGK IATVTLGSFG AATIDSSALT TVNLSGTGTS LGIGRGALTA TPTANTLTLN VNGLTTTGAI TDSEAA ADD GFTTINIAGS TASSTIASLV AADATTLNIS GDARVTITSH TAAALTGITV TNSVGATLGA ELATGLVFTG GAGADSI LL GATTKAIVMG AGDDTVTVSS ATLGAGGSVN GGDGTDVLVA NVNGSSFSAD PAFGGFETLR VAGAAAQGSH NANGFTAL Q LGATAGATTF TNVAVNVGLT VLAAPTGTTT VTLANATGTS DVFNLTLSSS AALAAGTVAL AGVETVNIAA TDTNTTAHV DTLTLQATSA KSIVVTGNAG LNLTNTGNTA VTSFDASAVT GTGSAVTFVS ANTTVGEVVT IRGGAGADSL TGSATANDTI IGGAGADTL VYTGGTDTFT GGTGADIFDI NAIGTSTAFV TITDAAVGDK LDLVGISTNG AIADGAFGAA VTLGAAATLA Q YLDAAAAG DGSGTSVAKW FQFGGDTYVV VDSSAGATFV SGADAVIKLT GLVTLTTSAF ATEVLTLA UniProtKB: S-layer protein rsaA |
-Macromolecule #3: CALCIUM ION
| Macromolecule | Name: CALCIUM ION / type: ligand / ID: 3 / Number of copies: 18 / Formula: CA |
|---|---|
| Molecular weight | Theoretical: 40.078 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | subtomogram averaging |
| Aggregation state | cell |
-Sample preparation
| Buffer | pH: 7 / Details: PYE medium |
|---|---|
| Grid | Model: Quantifoil R2/2 / Material: COPPER/RHODIUM / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 20 sec. / Pretreatment - Atmosphere: AIR / Details: 15 mA |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 283.15 K / Instrument: FEI VITROBOT MARK IV / Details: 1.5 s blot. |
| Details | Caulobacter crescentus stalk |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Specialist optics | Spherical aberration corrector: not used / Chromatic aberration corrector: not used / Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Frames/image: 1-10 / Number real images: 1 / Average exposure time: 1.0 sec. / Average electron dose: 3.4 e/Å2 / Details: Dose symmetric tilt scheme (Hagen et al, JSB) |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 5.0 µm / Calibrated defocus min: 2.0 µm / Calibrated magnification: 105000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 5.0 µm / Nominal defocus min: 2.0 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Final reconstruction | Number classes used: 3 / Applied symmetry - Point group: C6 (6 fold cyclic) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 3.5 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION / Number subtomograms used: 42990 |
|---|---|
| Extraction | Number tomograms: 110 / Number images used: 51866 / Reference model: EMDF / Method: EMD-10388 / Software - Name: RELION (ver. 4.0.0) / Details: RELION subtomogram averaging |
| Final 3D classification | Number classes: 6 / Avg.num./class: 8645 / Software - Name: RELION (ver. 4.0.0) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 4.0.0) / Details: RELION 4.0.0 |
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
|---|---|
| Output model | PDB-8bqe: |
