 ムービー
ムービー コントローラー
コントローラー 構造ビューア
構造ビューア 万見文献について
万見文献について



+検索条件
-Structure paper
| タイトル | IPET and FETR: experimental approach for studying molecular structure dynamics by cryo-electron tomography of a single-molecule structure. |
|---|---|
| ジャーナル・号・ページ | PLoS One, Vol. 7, Issue 1, Page e30249, Year 2012 |
| 掲載日 | 2012年1月24日 |
 著者 著者 | Lei Zhang / Gang Ren /  |
| PubMed 要旨 | The dynamic personalities and structural heterogeneity of proteins are essential for proper functioning. Structural determination of dynamic/heterogeneous proteins is limited by conventional ...The dynamic personalities and structural heterogeneity of proteins are essential for proper functioning. Structural determination of dynamic/heterogeneous proteins is limited by conventional approaches of X-ray and electron microscopy (EM) of single-particle reconstruction that require an average from thousands to millions different molecules. Cryo-electron tomography (cryoET) is an approach to determine three-dimensional (3D) reconstruction of a single and unique biological object such as bacteria and cells, by imaging the object from a series of tilting angles. However, cconventional reconstruction methods use large-size whole-micrographs that are limited by reconstruction resolution (lower than 20 Å), especially for small and low-symmetric molecule (<400 kDa). In this study, we demonstrated the adverse effects from image distortion and the measuring tilt-errors (including tilt-axis and tilt-angle errors) both play a major role in limiting the reconstruction resolution. Therefore, we developed a "focused electron tomography reconstruction" (FETR) algorithm to improve the resolution by decreasing the reconstructing image size so that it contains only a single-instance protein. FETR can tolerate certain levels of image-distortion and measuring tilt-errors, and can also precisely determine the translational parameters via an iterative refinement process that contains a series of automatically generated dynamic filters and masks. To describe this method, a set of simulated cryoET images was employed; to validate this approach, the real experimental images from negative-staining and cryoET were used. Since this approach can obtain the structure of a single-instance molecule/particle, we named it individual-particle electron tomography (IPET) as a new robust strategy/approach that does not require a pre-given initial model, class averaging of multiple molecules or an extended ordered lattice, but can tolerate small tilt-errors for high-resolution single "snapshot" molecule structure determination. Thus, FETR/IPET provides a completely new opportunity for a single-molecule structure determination, and could be used to study the dynamic character and equilibrium fluctuation of macromolecules. |
 リンク リンク | PLoS One / PubMed:22291925 / PubMed Central |
| 手法 | EM (サブトモグラム平均) |
| 解像度 | 14.1 - 42.0 Å |
| 構造データ | 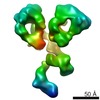 EMDB-2021: 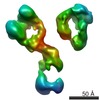 EMDB-2022:  EMDB-2023:  EMDB-2024: |
| 由来 |
|
 Homo sapiens (ヒト)
Homo sapiens (ヒト)