 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7490 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
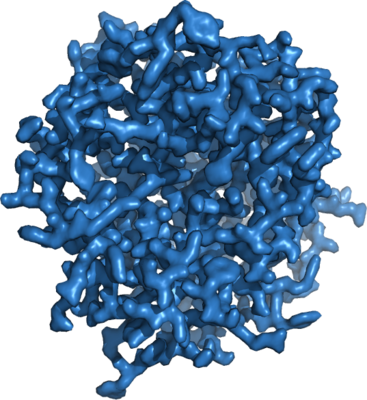
| Title | 1.71 A MicroED structure of proteinase K at 0.86 e- / A^2 | |||||||||
 Map data Map data | 1.71 A MicroED density map of proteinase K at 0.86 e- / A^2 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidase K / serine-type endopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Engyodontium album (fungus) / Parengyodontium album (fungus) Engyodontium album (fungus) / Parengyodontium album (fungus) | |||||||||
| Method | electron crystallography / cryo EM / Resolution: 1.71 Å | |||||||||
 Authors Authors | Hattne J / Shi D | |||||||||
 Citation Citation | Journal: Structure / Year: 2018 Title: Analysis of Global and Site-Specific Radiation Damage in Cryo-EM. Authors: Johan Hattne / Dan Shi / Calina Glynn / Chih-Te Zee / Marcus Gallagher-Jones / Michael W Martynowycz / Jose A Rodriguez / Tamir Gonen /  Abstract: Micro-crystal electron diffraction (MicroED) combines the efficiency of electron scattering with diffraction to allow structure determination from nano-sized crystalline samples in cryoelectron ...Micro-crystal electron diffraction (MicroED) combines the efficiency of electron scattering with diffraction to allow structure determination from nano-sized crystalline samples in cryoelectron microscopy (cryo-EM). It has been used to solve structures of a diverse set of biomolecules and materials, in some cases to sub-atomic resolution. However, little is known about the damaging effects of the electron beam on samples during such measurements. We assess global and site-specific damage from electron radiation on nanocrystals of proteinase K and of a prion hepta-peptide and find that the dynamics of electron-induced damage follow well-established trends observed in X-ray crystallography. Metal ions are perturbed, disulfide bonds are broken, and acidic side chains are decarboxylated while the diffracted intensities decay exponentially with increasing exposure. A better understanding of radiation damage in MicroED improves our assessment and processing of all types of cryo-EM data. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7490.map.gz | 2.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7490-v30.xmlemd-7490.xml | 13.7 KB 13.7 KB | Display Display | EMDB header |
| Images | 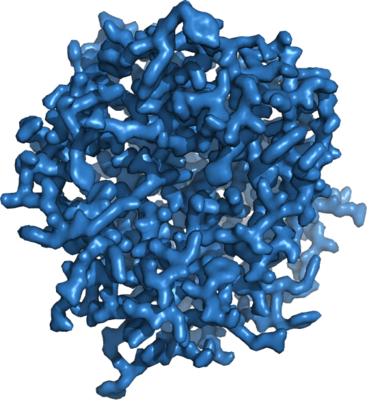 emd_7490.png emd_7490.png | 200.7 KB | ||
| Filedesc metadata | emd-7490.cif.gz | 5.3 KB | ||
| Filedesc structureFactors | emd_7490_sf.cif.gz | 1.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7490ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7490 http://ftp.pdbj.org/pub/emdb/structures/EMD-7490ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7490 | HTTPS FTP |
-Related structure data
| Related structure data |  6cl7MC  7491C  7492C  7493C  7494C  7495C  7496C  7497C  7498C  7499C  7500C  7501C  7502C  7503C  7504C  7505C  7506C  7507C  7508C  7509C  7510C  7511C  7512C  6cl8C  6cl9C  6claC  6clbC  6clcC  6cldC  6cleC  6clfC  6clgC  6clhC  6cliC  6cljC  6clkC  6cllC  6clmC  6clnC  6cloC  6clpC  6clqC  6clrC  6clsC  6cltC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_7490.map.gz / Format: CCP4 / Size: 3.2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 1.71 A MicroED density map of proteinase K at 0.86 e- / A^2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X: 0.55842 Å / Y: 0.55842 Å / Z: 0.57229 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
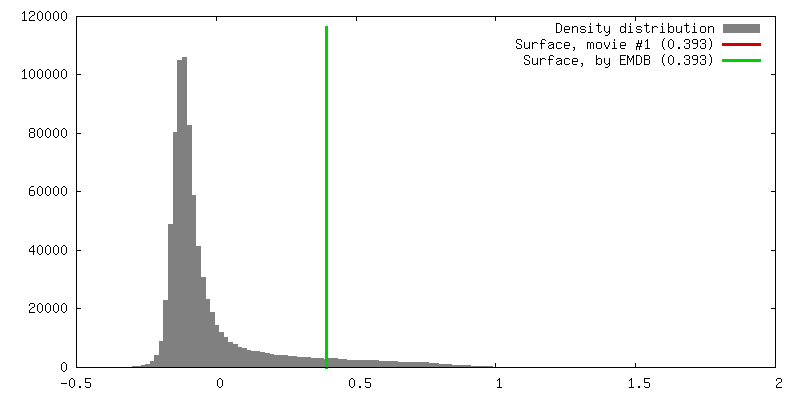
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 96 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) X (Row.)
X (Row.) Y (Col.)
Y (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Proteinase K
| Entire | Name: Proteinase K |
|---|---|
| Components |
|
-Supramolecule #1: Proteinase K
| Supramolecule | Name: Proteinase K / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Engyodontium album (fungus) |
| Molecular weight | Theoretical: 28.888994 KDa |
-Macromolecule #1: Proteinase K
| Macromolecule | Name: Proteinase K / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: peptidase K |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28.930783 KDa |
| Sequence | String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN ...String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN YSPASEPSVC TVGASDRYDR RSSFSNYGSV LDIFGPGTSI LSTWIGGSTR SISGTSMATP HVAGLAAYLM TL GKTTAAS ACRYIADTAN KGDLSNIPFG TVNLLAYNNY QA UniProtKB: Proteinase K |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | electron crystallography |
| Aggregation state | 3D array |
-Sample preparation
| Concentration | 25 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 30 % / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Image recording | Film or detector model: TVIPS TEMCAM-F416 (4k x 4k) / Digitization - Dimensions - Width: 2048 pixel / Digitization - Dimensions - Height: 2048 pixel / Number grids imaged: 1 / Number real images: 289 / Number diffraction images: 289 / Average exposure time: 5.1 sec. / Average electron dose: 0.0357 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: DIFFRACTION / Camera length: 1200 mm |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 1-279 / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | Electron scattering factors |
| Refinement | Space: RECIPROCAL / Protocol: OTHER / Overall B value: 6.613 |
| Output model | PDB-6cl7: |

