 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4990 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | T=4 MS2 Virus-like-particle | |||||||||
 Map data Map data | 0.04 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | MS2 / T=4 / BACTERIOPHAGE / VLP / VIRUS LIKE PARTICLE | |||||||||
| Function / homology | negative regulation of viral translation / Levivirus coat protein / Levivirus coat protein / Bacteriophage RNA-type, capsid / T=3 icosahedral viral capsid / structural molecule activity / RNA binding / identical protein binding / Capsid protein Function and homology information Function and homology information | |||||||||
| Biological species |  Escherichia phage MS2 (virus) / Escherichia virus MS2 Escherichia phage MS2 (virus) / Escherichia virus MS2 | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.0 Å | |||||||||
 Authors Authors | de Martin Garrido N / Ramlaul K | |||||||||
| Funding support |  United Kingdom, 2 items United Kingdom, 2 items
| |||||||||
 Citation Citation | Journal: Mol Microbiol / Year: 2020 Title: Bacteriophage MS2 displays unreported capsid variability assembling T = 4 and mixed capsids. Authors: Natàlia de Martín Garrido / Michael A Crone / Kailash Ramlaul / Paul A Simpson / Paul S Freemont / Christopher H S Aylett / Abstract: Bacteriophage MS2 is a positive-sense, single-stranded RNA virus encapsulated in an asymmetric T = 3 pseudo-icosahedral capsid. It infects Escherichia coli through the F-pilus, in which it binds ...Bacteriophage MS2 is a positive-sense, single-stranded RNA virus encapsulated in an asymmetric T = 3 pseudo-icosahedral capsid. It infects Escherichia coli through the F-pilus, in which it binds through a maturation protein incorporated into its capsid. Cryogenic electron microscopy has previously shown that its genome is highly ordered within virions, and that it regulates the assembly process of the capsid. In this study, we have assembled recombinant MS2 capsids with non-genomic RNA containing the capsid incorporation sequence, and investigated the structures formed, revealing that T = 3, T = 4 and mixed capsids between these two triangulation numbers are generated, and resolving structures of T = 3 and T = 4 capsids to 4 Å and 6 Å respectively. We conclude that the basic MS2 capsid can form a mix of T = 3 and T = 4 structures, supporting a role for the ordered genome in favouring the formation of functional T = 3 virions. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4990.map.gz | 190.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4990-v30.xmlemd-4990.xml | 18.1 KB 18.1 KB | Display Display | EMDB header |
| Images |  emd_4990.png emd_4990.png | 180.4 KB | ||
| Filedesc metadata | emd-4990.cif.gz | 5.9 KB | ||
| Others | emd_4990_half_map_1.map.gzemd_4990_half_map_2.map.gz | 165.9 MB 168.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4990ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4990 http://ftp.pdbj.org/pub/emdb/structures/EMD-4990ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4990 | HTTPS FTP |
-Related structure data
| Related structure data |  6rrtMC  4989C  6rrsC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_4990.map.gz / Format: CCP4 / Size: 216 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 0.04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.0277 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: MS2 bacteriophage T=4 virus-like particle half-map 1
| File | emd_4990_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | MS2 bacteriophage T=4 virus-like particle half-map 1 | ||||||||||||
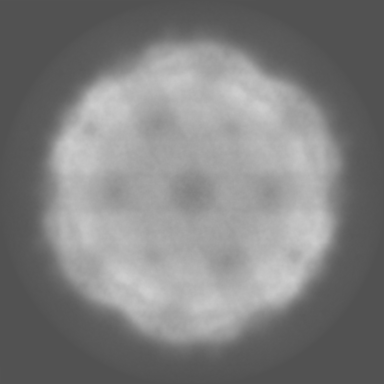
| Projections & Slices |
| ||||||||||||
| Density Histograms |



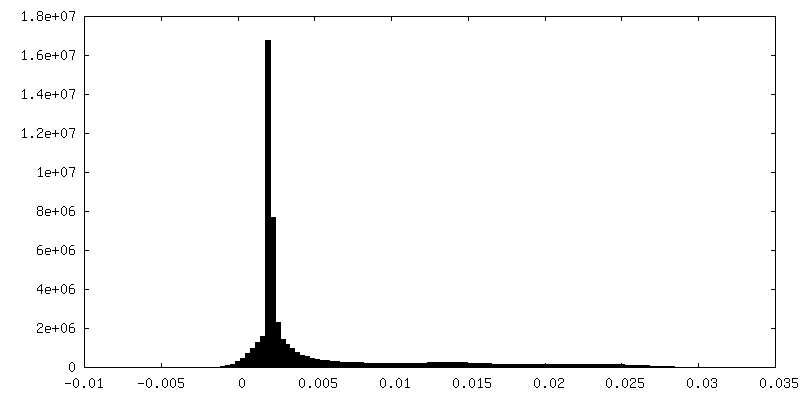
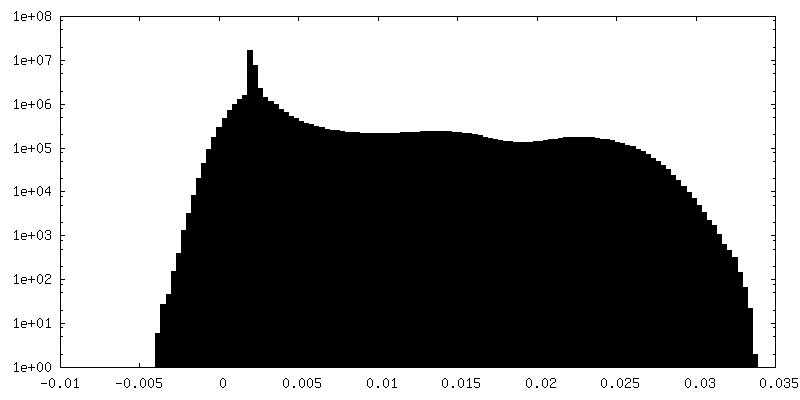
-Half map: MS2 bacteriophage T=4 virus-like particle half-map 2
| File | emd_4990_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | MS2 bacteriophage T=4 virus-like particle half-map 2 | ||||||||||||
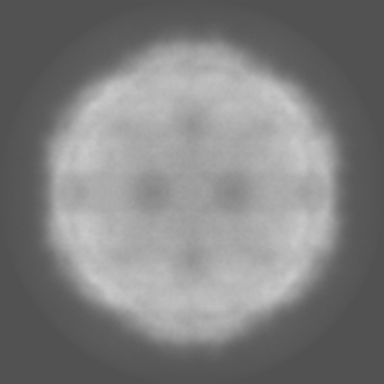
| Projections & Slices |
| ||||||||||||
| Density Histograms |


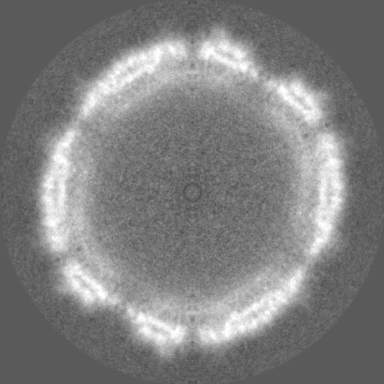

- Sample components
Sample components
-Entire : Escherichia virus MS2
| Entire | Name: Escherichia virus MS2 |
|---|---|
| Components |
|
-Supramolecule #1: Escherichia virus MS2
| Supramolecule | Name: Escherichia virus MS2 / type: virus / ID: 1 / Parent: 0 / Macromolecule list: all / NCBI-ID: 329852 / Sci species name: Escherichia virus MS2 / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: OTHER / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | Organism:  |
-Macromolecule #1: Capsid protein
| Macromolecule | Name: Capsid protein / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Escherichia phage MS2 (virus) |
| Molecular weight | Theoretical: 13.869659 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MASNFTQFVL VDNGGTGDVT VAPSNFANGV AEWISSNSRS QAYKVTCSVR QSSAQNRKYT IKVEVPKVAT QTVGGVELPV AAWRSYLNM ELTIPIFATN SDCELIVKAM QGLLKDGNPI PSAIAANSGI Y UniProtKB: Capsid protein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 / Details: 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 100 mM NaCl |
|---|---|
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: GRAPHENE OXIDE / Support film - topology: HOLEY ARRAY / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Time: 15 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 278 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Image recording | Film or detector model: FEI FALCON I (4k x 4k) / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 4 / Number real images: 2129 / Average exposure time: 4.0 sec. / Average electron dose: 80.0 e/Å2 Details: Frames were obtained with a beam-splitter Images were recorded at both 100kx and 150kx and resampled in Fourier space to 100kx for refinement |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: -2.0 µm / Nominal defocus min: -0.3 µm / Nominal magnification: 100000 |
| Sample stage | Specimen holder model: GATAN 626 SINGLE TILT LIQUID NITROGEN CRYO TRANSFER HOLDER Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |


