Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-4625: E. coli ClpB (DWB and K476C mutant) bound to casein - state KC-2 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4625 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | E. coli ClpB (DWB and K476C mutant) bound to casein - state KC-2 | ||||||||||||
 Map data Map data | ClpB-DWB-K476C bound to casein in presence of ATPgammaS - state KC-2 | ||||||||||||
 Sample Sample |
| ||||||||||||
| Biological species |   | ||||||||||||
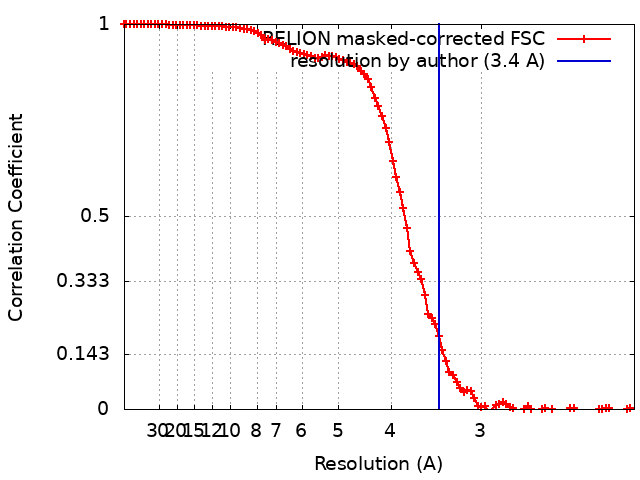
| Method | single particle reconstruction / cryo EM / Resolution: 3.4 Å | ||||||||||||
 Authors Authors | Deville C / Saibil HR | ||||||||||||
| Funding support |  United Kingdom, 3 items United Kingdom, 3 items
| ||||||||||||
 Citation Citation | Journal: Cell Rep / Year: 2019 Title: Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor. Authors: Célia Deville / Kamila Franke / Axel Mogk / Bernd Bukau / Helen R Saibil /  Abstract: AAA+ proteins form asymmetric hexameric rings that hydrolyze ATP and thread substrate proteins through a central channel via mobile substrate-binding pore loops. Understanding how ATPase and ...AAA+ proteins form asymmetric hexameric rings that hydrolyze ATP and thread substrate proteins through a central channel via mobile substrate-binding pore loops. Understanding how ATPase and threading activities are regulated and intertwined is key to understanding the AAA+ protein mechanism. We studied the disaggregase ClpB, which contains tandem ATPase domains (AAA1, AAA2) and shifts between low and high ATPase and threading activities. Coiled-coil M-domains repress ClpB activity by encircling the AAA1 ring. Here, we determine the mechanism of ClpB activation by comparing ATPase mechanisms and cryo-EM structures of ClpB wild-type and a constitutively active ClpB M-domain mutant. We show that ClpB activation reduces ATPase cooperativity and induces a sequential mode of ATP hydrolysis in the AAA2 ring, the main ATPase motor. AAA1 and AAA2 rings do not work synchronously but in alternating cycles. This ensures high grip, enabling substrate threading via a processive, rope-climbing mechanism. | ||||||||||||
| History |
|
- Structure visualization
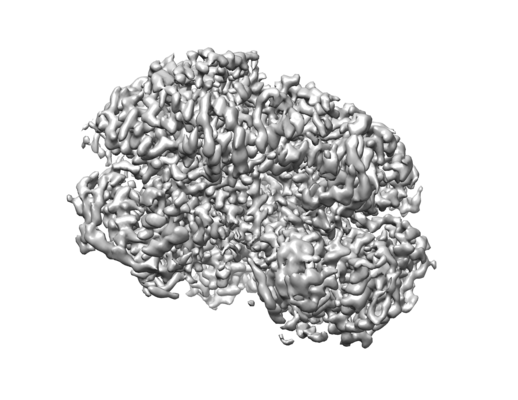
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4625.map.gz | 85.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4625-v30.xmlemd-4625.xml | 20.8 KB 20.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_4625_fsc.xml | 10.3 KB | Display | FSC data file |
| Images | 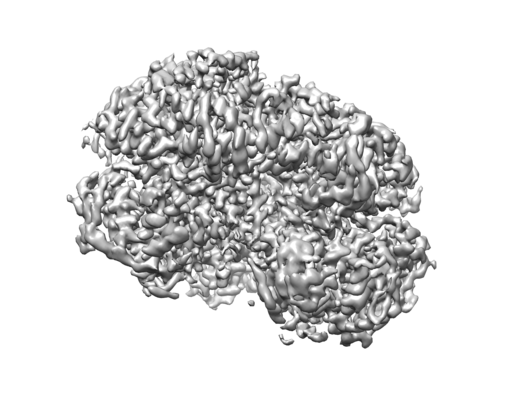 emd_4625.png emd_4625.png | 88.2 KB | ||
| Masks | emd_4625_msk_1.map | 91.1 MB | Mask map | |
| Others | emd_4625_half_map_1.map.gzemd_4625_half_map_2.map.gz | 29.7 MB 29.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4625ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4625 http://ftp.pdbj.org/pub/emdb/structures/EMD-4625ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4625 | HTTPS FTP |
-Validation report
| Summary document | emd_4625_validation.pdf.gz | 402.2 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_4625_full_validation.pdf.gz | 401.4 KB | Display | |
| Data in XML | emd_4625_validation.xml.gz | 15.9 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-4625ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-4625 | HTTPS FTP |
-Related structure data
| Related structure data |  4621C  4622C  4623C  4624C  4626C  4627C  4940C  4941C  4942C  6qs4C  6qs6C  6qs7C  6qs8C  6rn2C  6rn3C  6rn4C C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_4625.map.gz / Format: CCP4 / Size: 91.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | ClpB-DWB-K476C bound to casein in presence of ATPgammaS - state KC-2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
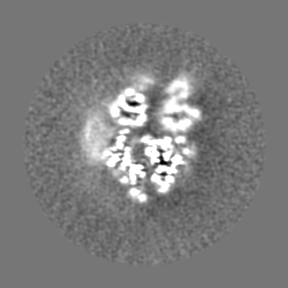
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.05 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
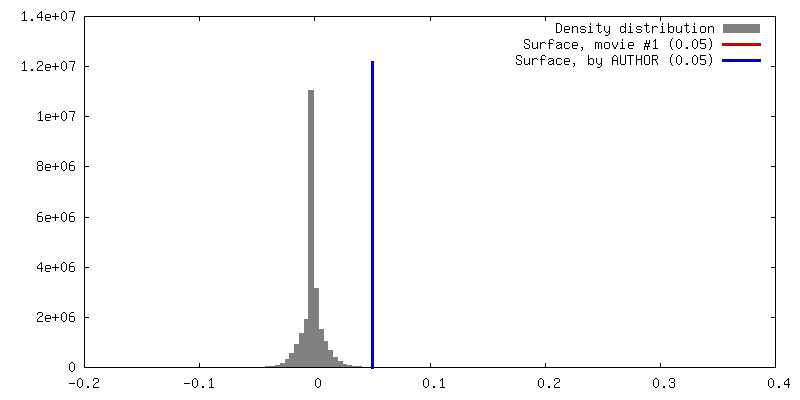
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_4625_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
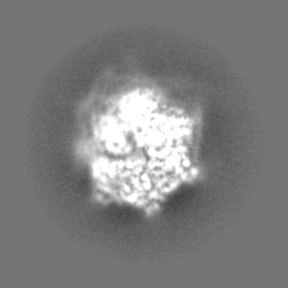
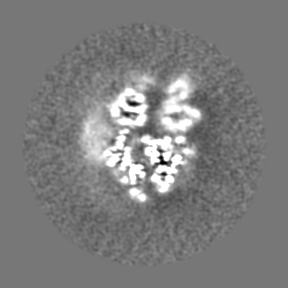
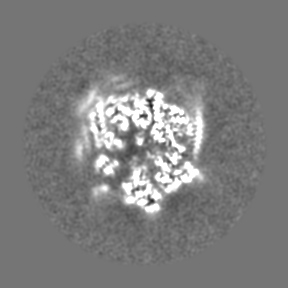
| Projections & Slices |
| ||||||||||||
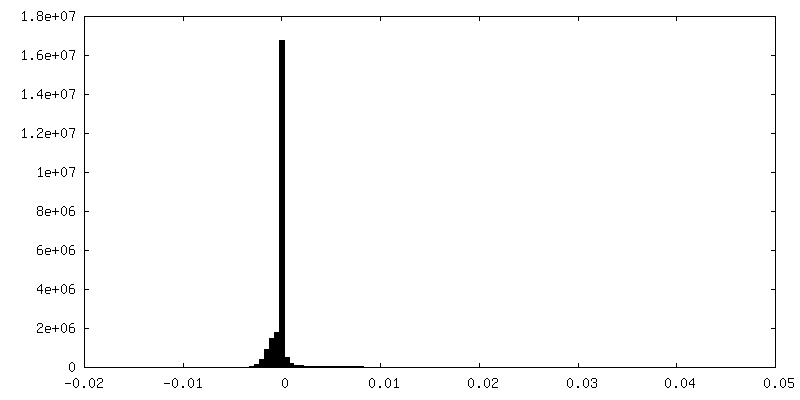
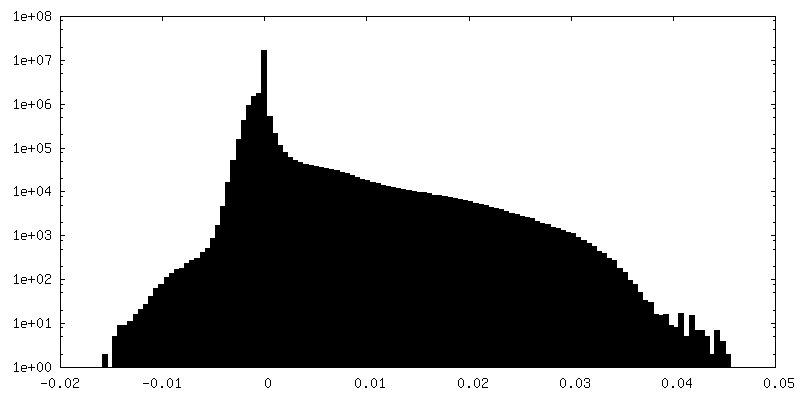
| Density Histograms |






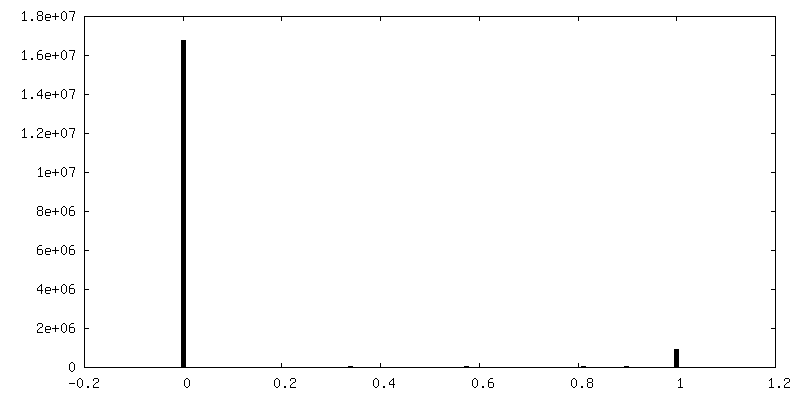
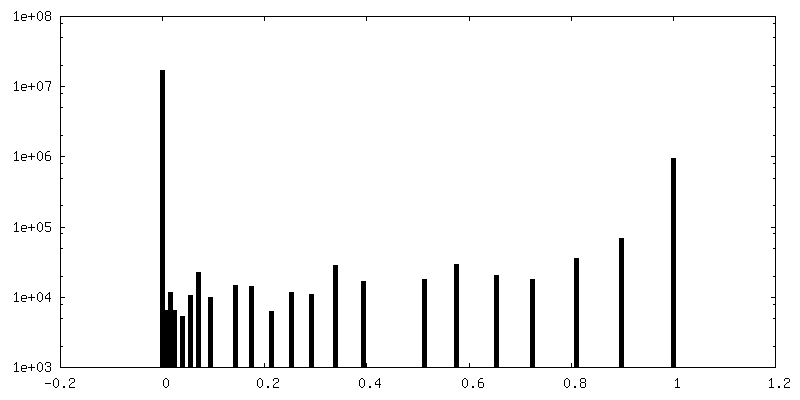
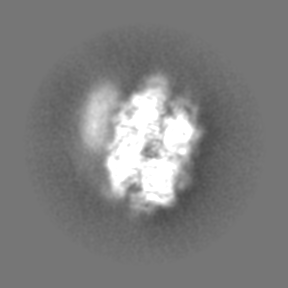
-Half map: Half map 1
| File | emd_4625_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: Half map 2
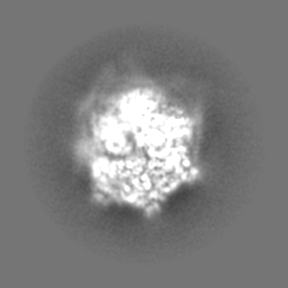
| File | emd_4625_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : ClpB-DWB-K476C bound to casein in presence of ATPgammaS
| Entire | Name: ClpB-DWB-K476C bound to casein in presence of ATPgammaS |
|---|---|
| Components |
|
-Supramolecule #1: ClpB-DWB-K476C bound to casein in presence of ATPgammaS
| Supramolecule | Name: ClpB-DWB-K476C bound to casein in presence of ATPgammaS type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Molecular weight | Theoretical: 570 KDa |
-Macromolecule #1: ClpB
| Macromolecule | Name: ClpB / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Sequence | String: MRLDRLTNKF QLALADAQSL ALGHDNQFIE PLHLMSALLN QEGGSVSPLL TSAGINAGQL RTDINQALN RLPQVEGTGG DVQPSQDLVR VLNLCDKLAQ KRGDNFISSE LFVLAALESR G TLADILKA AGATTANITQ AIEQMRGGES VNDQGAEDQR QALKKYTIDL ...String: MRLDRLTNKF QLALADAQSL ALGHDNQFIE PLHLMSALLN QEGGSVSPLL TSAGINAGQL RTDINQALN RLPQVEGTGG DVQPSQDLVR VLNLCDKLAQ KRGDNFISSE LFVLAALESR G TLADILKA AGATTANITQ AIEQMRGGES VNDQGAEDQR QALKKYTIDL TERAEQGKLD PV IGRDEEI RRTIQVLQRR TKNNPVLIGE PGVGKTAIVE GLAQRIINGE VPEGLKGRRV LAL DMGALV AGAKYRGEFE ERLKGVLNDL AKQEGNVILF IDALHTMVGA GKADGAMDAG NMLK PALAR GELHCVGATT LDEYRQYIEK DAALERRFQK VFVAEPSVED TIAILRGLKE RYELH HHVQ ITDPAIVAAA TLSHRYIADR QLPDKAIDLI DEAASSIRMQ IDSKPEELDR LDRRII QLK LEQQALMKES DEASKKRLDM LNEELSDKER QYSELEEEWK AEKASLSGTQ TICAELE QA KIAIEQARRV GDLARMSELQ YGKIPELEKQ LEAATQLEGK TMRLLRNKVT DAEIAEVL A RWTGIPVSRM MESEREKLLR MEQELHHRVI GQNEAVDAVS NAIRRSRAGL ADPNRPIGS FLFLGPTGVG KTELCKALAN FMFDSDEAMV RIDMSEFMEK HSVSRLVGAP PGYVGYEEGG YLTEAVRRR PYSVILLDAV EKAHPDVFNI LLQVLDDGRL TDGQGRTVDF RNTVVIMTSN L GSDLIQER FGELDYAHMK ELVLGVVSHN FRPEFINRID EVVVFHPLGE QHIASIAQIQ LK RLYKRLE ERGYEIHISD EALKLLSENG YDPVYGARPL KRAIQQQIEN PLAQQILSGE LVP GKVIRL EVNEDRIVAV Q |
-Macromolecule #2: casein
| Macromolecule | Name: casein / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Sequence | String: MKLLILTCLV AVALARPKHP IKHQGLPQEV LNENLLRFFV APFPEVFGKE KVNELSKDIG SESTEDQAM EDIKQMEAES ISSSEEIVPN SVEQKHIQKE DVPSERYLGY LEQLLRLKKY K VPQLEIVP NSAEERLHSM KEGIHAQQKE PMIGVNQELA YFYPELFRQF ...String: MKLLILTCLV AVALARPKHP IKHQGLPQEV LNENLLRFFV APFPEVFGKE KVNELSKDIG SESTEDQAM EDIKQMEAES ISSSEEIVPN SVEQKHIQKE DVPSERYLGY LEQLLRLKKY K VPQLEIVP NSAEERLHSM KEGIHAQQKE PMIGVNQELA YFYPELFRQF YQLDAYPSGA WY YVPLGTQ YTDAPSFSDI PNPIGSENSE KTTMPLW |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.6 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||||||||
| Grid | Model: Quantifoil, UltrAuFoil / Material: GOLD / Mesh: 300 / Support film - Material: GRAPHENE OXIDE / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR | ||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 295 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 6060 / Average electron dose: 1.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.5 µm / Nominal defocus min: 1.5 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |