 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3782: A 3.4 Angstrom structure of HIV-1 CA-SP1 by 3D-CTF correction of ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3782 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | A 3.4 Angstrom structure of HIV-1 CA-SP1 by 3D-CTF correction of cryo-electron tomograms | |||||||||
 Map data Map data | None | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | |||||||||
| Method | subtomogram averaging / cryo EM / Resolution: 3.4 Å | |||||||||
 Authors Authors | Turonova B / Schur FKM / Wan W / Briggs JAG | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2017 Title: Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4Å. Authors: Beata Turoňová / Florian K M Schur / William Wan / John A G Briggs /   Abstract: Cryo-electron tomography (cryo-ET) allows cellular ultrastructures and macromolecular complexes to be imaged in three-dimensions in their native environments. Cryo-electron tomograms are ...Cryo-electron tomography (cryo-ET) allows cellular ultrastructures and macromolecular complexes to be imaged in three-dimensions in their native environments. Cryo-electron tomograms are reconstructed from projection images taken at defined tilt-angles. In order to recover high-resolution information from cryo-electron tomograms, it is necessary to measure and correct for the contrast transfer function (CTF) of the microscope. Most commonly, this is performed using protocols that approximate the sample as a two-dimensional (2D) plane. This approximation accounts for differences in defocus and therefore CTF across the tilted sample. It does not account for differences in defocus of objects at different heights within the sample; instead, a 3D approach is required. Currently available approaches for 3D-CTF correction are computationally expensive and have not been widely implemented. Here we simulate the benefits of 3D-CTF correction for high-resolution subtomogram averaging, and present a user-friendly, computationally-efficient 3D-CTF correction tool, NovaCTF, that is compatible with standard tomogram reconstruction workflows in IMOD. We validate the approach on synthetic data and test it using subtomogram averaging of real data. Consistent with our simulations, we find that 3D-CTF correction allows high-resolution structures to be obtained with much smaller subtomogram averaging datasets than are required using 2D-CTF. We also show that using equivalent dataset sizes, 3D-CTF correction can be used to obtain higher-resolution structures. We present a 3.4Å resolution structure determined by subtomogram averaging. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
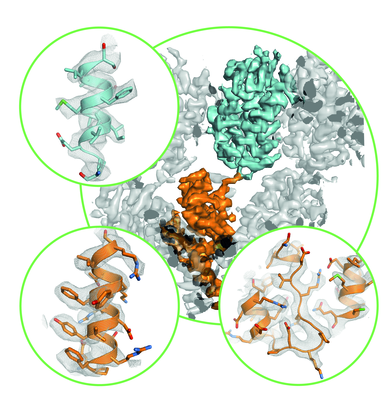
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3782.map.gz | 24.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3782-v30.xmlemd-3782.xml | 14.4 KB 14.4 KB | Display Display | EMDB header |
| Images | 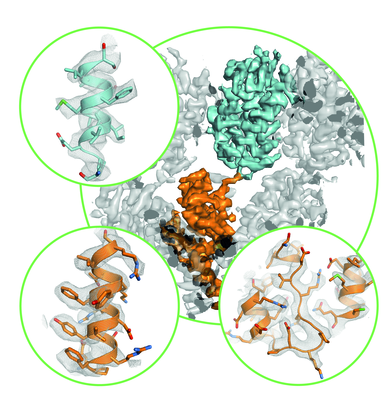 emd_3782.png emd_3782.png | 694.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3782ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3782 http://ftp.pdbj.org/pub/emdb/structures/EMD-3782ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3782 | HTTPS FTP |
-Validation report
| Summary document | emd_3782_validation.pdf.gz | 317 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_3782_full_validation.pdf.gz | 316.1 KB | Display | |
| Data in XML | emd_3782_validation.xml.gz | 5.8 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-3782ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-3782 | HTTPS FTP |
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10164 (Title: Cryo-electron tomography of immature HIV-1 dMACANC VLPs Data size: 865.0 Data #1: Compressed, unaligned, multi-frame micrographs of tilt series containing HIV-1 dMACANC virus like particles assembled in the presence of BVM. [micrographs - multiframe]) |







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_3782.map.gz / Format: CCP4 / Size: 27 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Human immunodeficiency virus 1
| Entire | Name: Human immunodeficiency virus 1 |
|---|---|
| Components |
|
-Supramolecule #1: Human immunodeficiency virus 1
| Supramolecule | Name: Human immunodeficiency virus 1 / type: virus / ID: 1 / Parent: 0 / Macromolecule list: #1 Details: Virus-like particles were obtained by in vitro assembly of a truncated Gag construct (deltaMACANCSP2) in presence of the maturation inhibitor Bevirimat NCBI-ID: 11676 / Sci species name: Human immunodeficiency virus 1 / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: OTHER / Virus enveloped: No / Virus empty: Yes |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Host system | Organism:  |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | subtomogram averaging |
| Aggregation state | particle |
-Sample preparation
| Concentration | 5 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
Details: Virus-like particles were assembled in the presence of nucleic acid (73mer oligonucleotide, 1:10 molar ratio oligonucleotide:protein). | |||||||||||||||
| Grid | Model: C-flat / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Details: at 20 mA | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 288 K / Instrument: FEI VITROBOT MARK II Details: 10nM colloidal gold was added to the sample prior to plunge freezing.. | |||||||||||||||
| Details | Virus-like particles were assembled in vitro |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Lower energy threshold: 0 eV / Energy filter - Upper energy threshold: 20 eV |
| Details | Nanoprobe |
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Frames/image: 8-10 / Number grids imaged: 2 / Average exposure time: 1.0 sec. / Average electron dose: 3.4 e/Å2 Details: Number of frames ranged from 8-10 Exposure time per tilt ranged from 0.8 to 1.0 seconds |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 5.0 µm / Nominal defocus min: 1.5 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
