ジャーナル: Protein Sci / 年: 2023 タイトル: Regulatory mechanisms of c-di-AMP synthase from Mycobacterium smegmatis revealed by a structure: Function analysis. 著者: Sudhanshu Gautam / Avisek Mahapa / Lahari Yeramala / Apoorv Gandhi / Sushma Krishnan / Vinothkumar Kutti R / Dipankar Chatterji / 要旨: Cyclic-di-nucleotide-based secondary messengers regulate various physiological functions, including stress responses in bacteria. Cyclic diadenosine monophosphate (c-di-AMP) has recently emerged as a ...Cyclic-di-nucleotide-based secondary messengers regulate various physiological functions, including stress responses in bacteria. Cyclic diadenosine monophosphate (c-di-AMP) has recently emerged as a crucial second messenger with implications in processes including osmoregulation, antibiotic resistance, biofilm formation, virulence, DNA repair, ion homeostasis, and sporulation, and has potential therapeutic applications. The contrasting activities of the enzymes diadenylate cyclase (DAC) and phosphodiesterase (PDE) determine the equilibrium levels of c-di-AMP. Although c-di-AMP is suspected of playing an essential role in the pathophysiology of bacterial infections and in regulating host-pathogen interactions, the mechanisms of its regulation remain relatively unexplored in mycobacteria. In this report, we biochemically and structurally characterize the c-di-AMP synthase (MsDisA) from Mycobacterium smegmatis. The enzyme activity is regulated by pH and substrate concentration; conditions of significance in the homoeostasis of c-di-AMP levels. Substrate binding stimulates conformational changes in the protein, and pApA and ppApA are synthetic intermediates detectable when enzyme efficiency is low. Unlike the orthologous Bacillus subtilis enzyme, MsDisA does not bind to, and its activity is not influenced in the presence of DNA. Furthermore, we have determined the cryo-EM structure of MsDisA, revealing asymmetry in its structure in contrast to the symmetric crystal structure of Thermotoga maritima DisA. We also demonstrate that the N-terminal minimal region alone is sufficient and essential for oligomerization and catalytic activity. Our data shed light on the regulation of mycobacterial DisA and possible future directions to pursue.
選択した数: 973755 詳細: Template Picker from cryoSparc 3.0 were used to pick total particles in the first extraction. Three rounds of reference-free 2d classification were carried to remove bad particles followed by NU-refinement.
初期モデル
モデルのタイプ: OTHER / 詳細: Initial model was obtained with cryosparc 3.0.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報
 マップデータ
マップデータ 試料
試料 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Mycolicibacterium smegmatis MC2 155 (バクテリア)
Mycolicibacterium smegmatis MC2 155 (バクテリア) データ登録者
データ登録者 インド, 2件
インド, 2件  引用
引用 構造の表示
構造の表示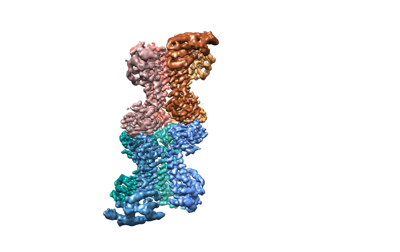
 ダウンロードとリンク
ダウンロードとリンク emd_33540.png
emd_33540.png http://ftp.pdbj.org/pub/emdb/structures/EMD-33540
http://ftp.pdbj.org/pub/emdb/structures/EMD-33540
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




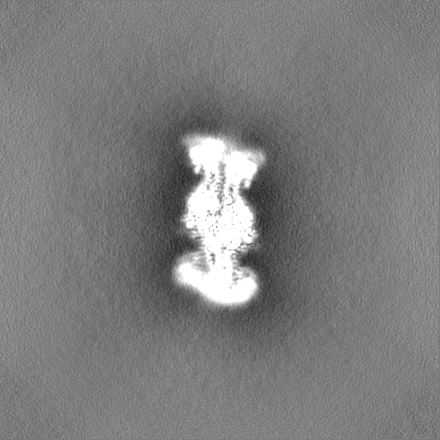

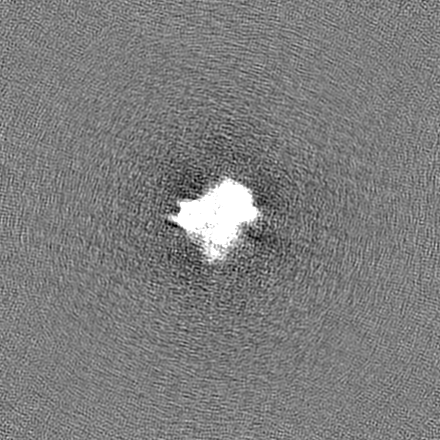



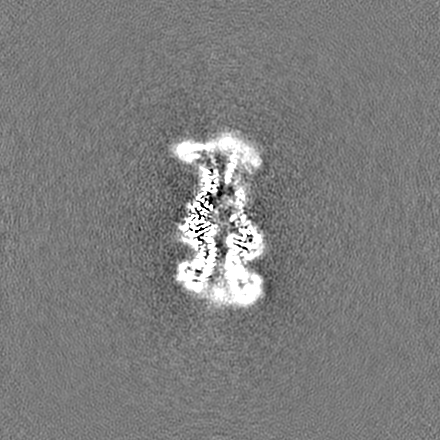
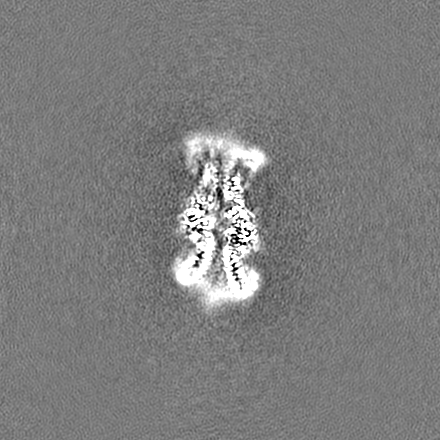

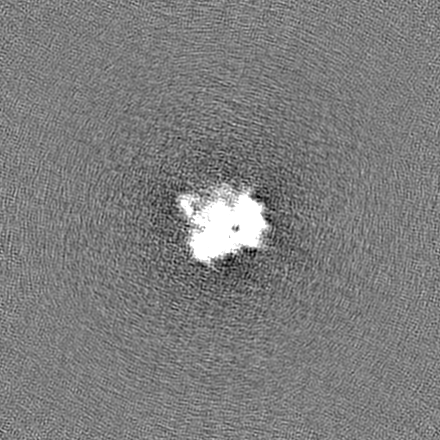



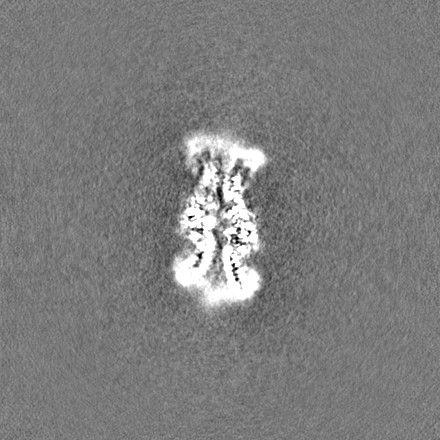
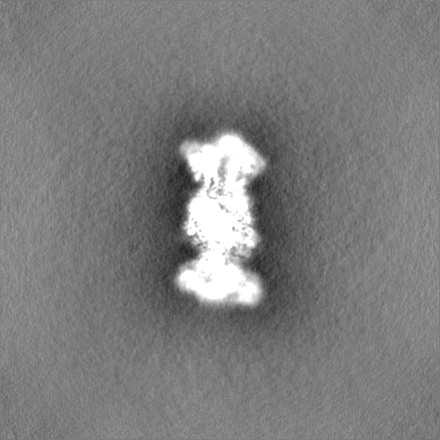
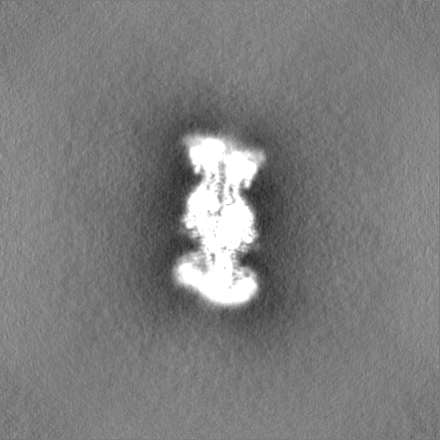

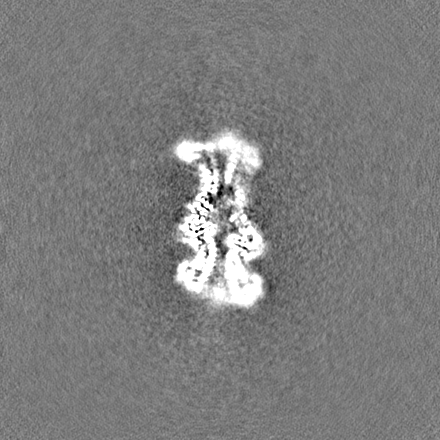
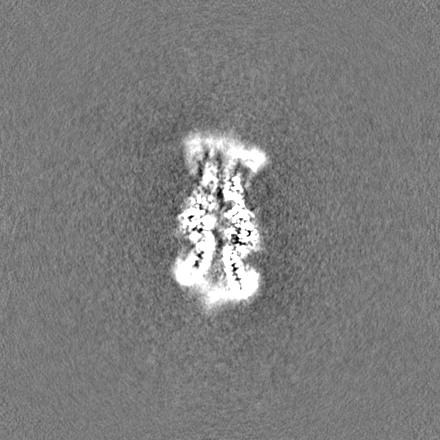
 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN