 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-33350: Cryo-EM structure of double occupied ring (DOR) of GroEL-UGT1A co... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of double occupied ring (DOR) of GroEL-UGT1A complex at 2.7 Ang. resolution | ||||||||||||||||||||||||
 Map data Map data | 2.7 Ang. Resolution Double Occupied Ring (DOR) GroEL-UGT1A Structure Complex | ||||||||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||||||||
 Keywords Keywords | cryogenic electron microscopy / single-particle analysis / molecular motion / structure-function relationship / focus classification / separating heterogeneity / groel / chaperone / unfolded protein | ||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationGroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding ...GroEL-GroES complex / chaperonin ATPase / virion assembly / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / : / response to heat / protein refolding / protein folding / magnesium ion binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / cytosol Similarity search - Function | ||||||||||||||||||||||||
| Biological species |   Homo sapiens (human) Homo sapiens (human) | ||||||||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.7 Å | ||||||||||||||||||||||||
 Authors Authors | Stapleton K / Takagi J / Mizohata E | ||||||||||||||||||||||||
| Funding support |  Japan, 7 items Japan, 7 items
| ||||||||||||||||||||||||
 Citation Citation | Journal: To Be Published Title: Unmasking GroEL: Structure, dynamics, and substrate binding revealed by single-particle cryo-EM Authors: Stapleton KM / Mizobata T / Miyazaki N / Takatsuji T / Kato T / Iwasaki K / Standley DM / Kawamura T / Nakane T / Takagi J / Mizohata E | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
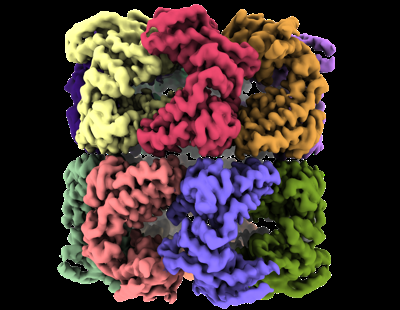
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_33350.map.gz | 79.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-33350-v30.xmlemd-33350.xml | 21.5 KB 21.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_33350_fsc.xml | 10.7 KB | Display | FSC data file |
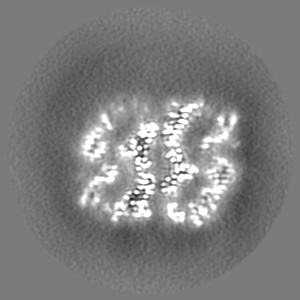
| Images | 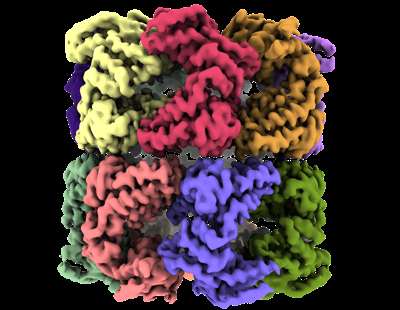 emd_33350.png emd_33350.png | 141.9 KB | ||
| Masks | emd_33350_msk_1.map | 103 MB | Mask map | |
| Filedesc metadata | emd-33350.cif.gz | 7.3 KB | ||
| Others | emd_33350_half_map_1.map.gzemd_33350_half_map_2.map.gz | 80.4 MB 80.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-33350ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33350 http://ftp.pdbj.org/pub/emdb/structures/EMD-33350ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33350 | HTTPS FTP |
-Related structure data
| Related structure data |  7xokMC  7xojC  7xolC  7xomC  7xonC  7xooC  7xopC  7xoqC  7xorC  7xosC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_33350.map.gz / Format: CCP4 / Size: 103 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 2.7 Ang. Resolution Double Occupied Ring (DOR) GroEL-UGT1A Structure Complex | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.87 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_33350_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
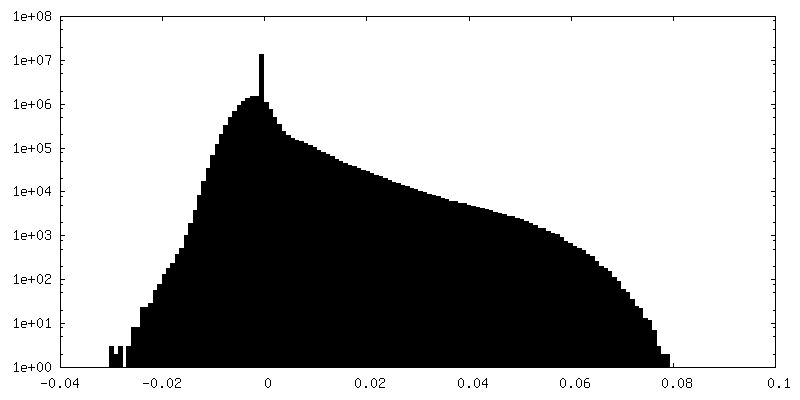
| Density Histograms |





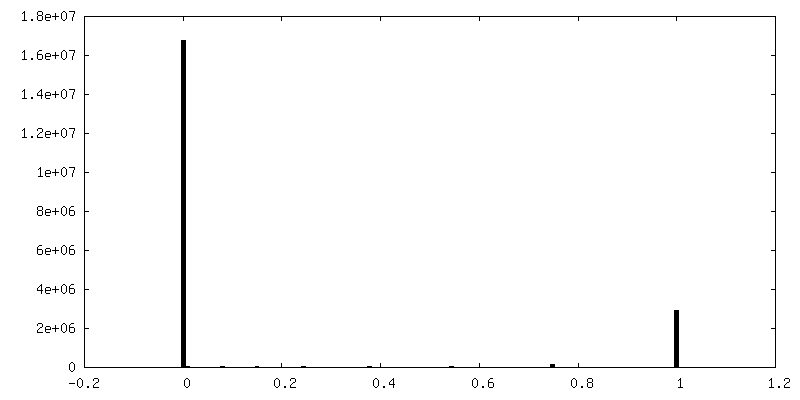
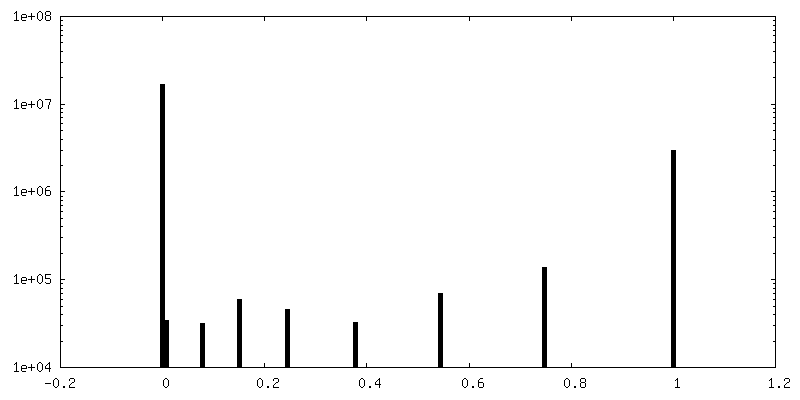
-Half map: half map-2 2.7 Ang. Resolution Double Occupied Ring (DOR) GroEL-UGT1A...
| File | emd_33350_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half_map-2__2.7 Ang. Resolution Double Occupied Ring (DOR) GroEL-UGT1A Structure Complex | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

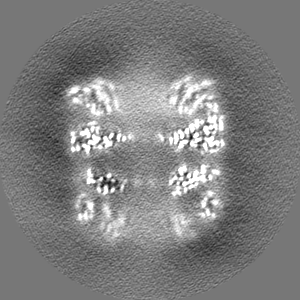
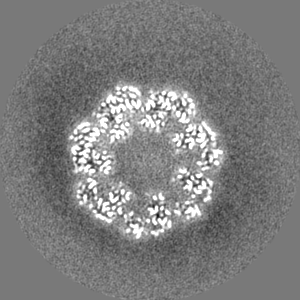
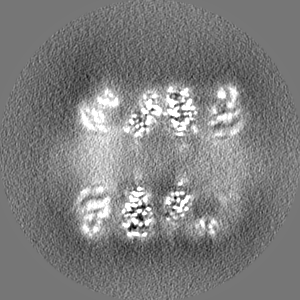
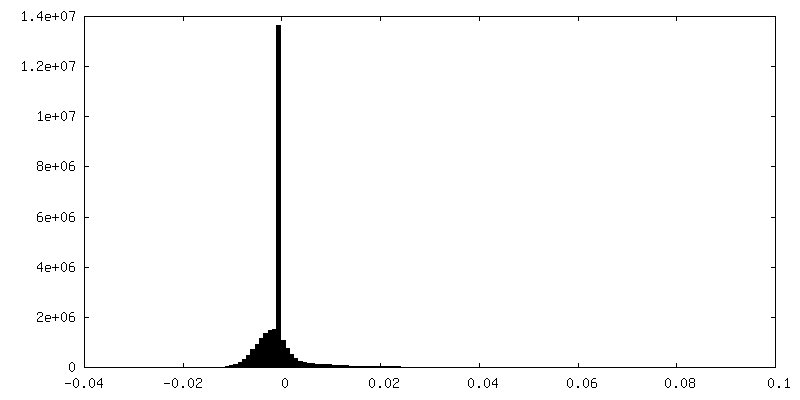
-Half map: half map-1 2.7 Ang. Resolution Double Occupied Ring (DOR) GroEL-UGT1A...
| File | emd_33350_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half_map-1__2.7 Ang. Resolution Double Occupied Ring (DOR) GroEL-UGT1A Structure Complex | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : Double occupied ring (DOR) of GroEL-UGT1A complex
| Entire | Name: Double occupied ring (DOR) of GroEL-UGT1A complex |
|---|---|
| Components |
|
-Supramolecule #1: Double occupied ring (DOR) of GroEL-UGT1A complex
| Supramolecule | Name: Double occupied ring (DOR) of GroEL-UGT1A complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: GroEL complexed with unfolded UGT1A present in two rings |
|---|---|
| Source (natural) | Organism: |
-Supramolecule #2: GroEL
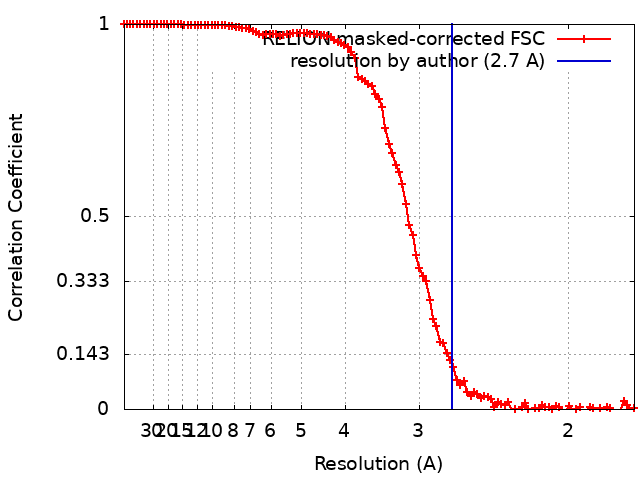
| Supramolecule | Name: GroEL / type: complex / ID: 2 / Parent: 1 / Macromolecule list: all Details: The model was built in a map at 2.7 Ang. global resolution by FSC 0.143 |
|---|---|
| Source (natural) | Organism: |
-Supramolecule #3: UDP-glucuronosyltransferase 1A (UGT1A)
| Supramolecule | Name: UDP-glucuronosyltransferase 1A (UGT1A) / type: complex / ID: 3 / Parent: 1 / Macromolecule list: all / Details: Model was not built because of weak density map |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
-Macromolecule #1: Chaperonin GroEL
| Macromolecule | Name: Chaperonin GroEL / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO / EC number: chaperonin ATPase |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 57.260504 KDa |
| Recombinant expression | Organism: |
| Sequence | String: AAKDVKFGND ARVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTAAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV ...String: AAKDVKFGND ARVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTAAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV GKEGVITVED GTGLQDELDV VEGMQFDRGY LSPYFINKPE TGAVELESPF ILLADKKISN IREMLPVLEA VA KAGKPLL IIAEDVEGEA LATLVVNTMR GIVKVAAVKA PGFGDRRKAM LQDIATLTGG TVISEEIGME LEKATLEDLG QAK RVVINK DTTTIIDGVG EEAAIQGRVA QIRQQIEEAT SDYDREKLQE RVAKLAGGVA VIKVGAATEV EMKEKKARVE DALH ATRAA VEEGVVAGGG VALIRVASKL ADLRGQNEDQ NVGIKVALRA MEAPLRQIVL NCGEEPSVVA NTVKGGDGNY GYNAA TEEY GNMIDMGILD PTKVTRSALQ YAASVAGLMI TTECMVTDLP KNDAADLGAA GGMGGMGGMG GMM UniProtKB: Chaperonin GroEL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 33 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
Details: Sample containing GorEL-UGT1A was made fresh and used without undergoing any freeze-thaw cycles to avoid degradation in the solution. The sample was in a buffer solution of 150mM NaCl, 20mM Tris-HCl at pH 7.5 | |||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 90 sec. / Pretreatment - Atmosphere: AIR | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV Details: 3 ul of 33 mg/ml GroEL-UGT1A was placed on Holey carbon Quanitifoil copper grids (300 mesh size R1.2/1.3) and blotted for 3 seconds (blot force =1). |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 2951 / Average electron dose: 40.0 e/Å2 Details: Using a Titan KRIOS TEM operated at 300 kV, 2,951 movies were collected using the Falcon III DED in Counting mode at a magnification of 75000x corresponding to a pixel size of 0.87 Pixel/A ...Details: Using a Titan KRIOS TEM operated at 300 kV, 2,951 movies were collected using the Falcon III DED in Counting mode at a magnification of 75000x corresponding to a pixel size of 0.87 Pixel/A at the specimen level. All 2,951 movies were imported into the RELION pipeline and prepared for single-particle analysis |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.2 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Overall B value: 76.71 |
|---|---|
| Output model | PDB-7xok: |
