 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-24799: Rabbit muscle aldolase determined in the presence of 20% v/v glycerol. -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-24799 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Rabbit muscle aldolase determined in the presence of 20% v/v glycerol. | |||||||||
 Map data Map data | Rabbit muscle aldolase determined in the presence of 20% v/v glycerol. | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of Arp2/3 complex-mediated actin nucleation / fructose-bisphosphate aldolase / fructose-bisphosphate aldolase activity / I band / M band / glycolytic process / protein homotetramerization / positive regulation of cell migration Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.6 Å | |||||||||
 Authors Authors | Basanta B / Hirschi M / Lander GC | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2022 Title: A case for glycerol as an acceptable additive for single-particle cryoEM samples. Authors: Benjamin Basanta / Marscha M Hirschi / Danielle A Grotjahn / Gabriel C Lander / Abstract: Buffer-composition and sample-preparation guidelines for cryo-electron microscopy are geared towards maximizing imaging contrast and reducing electron-beam-induced motion. These pursuits often ...Buffer-composition and sample-preparation guidelines for cryo-electron microscopy are geared towards maximizing imaging contrast and reducing electron-beam-induced motion. These pursuits often involve the minimization or the complete removal of additives that are commonly used to facilitate proper protein folding and minimize aggregation. Among these admonished additives is glycerol, a widely used osmolyte that aids protein stability. In this work, it is shown that the inclusion of glycerol does not preclude high-resolution structure determination by cryoEM, as demonstrated by an ∼2.3 Å resolution reconstruction of mouse apoferritin (∼500 kDa) and an ∼3.3 Å resolution reconstruction of rabbit muscle aldolase (∼160 kDa) in the presence of 20%(v/v) glycerol. While it was found that generating thin ice that is amenable to high-resolution imaging requires long blot times, the addition of glycerol did not result in increased beam-induced motion or an inability to pick particles. Overall, these findings indicate that glycerol should not be discounted as a cryoEM sample-buffer additive, particularly for large, fragile complexes that are prone to disassembly or aggregation upon its removal. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_24799.map.gz | 6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-24799-v30.xmlemd-24799.xml | 18.5 KB 18.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_24799_fsc.xml | 9.2 KB | Display | FSC data file |
| Images | 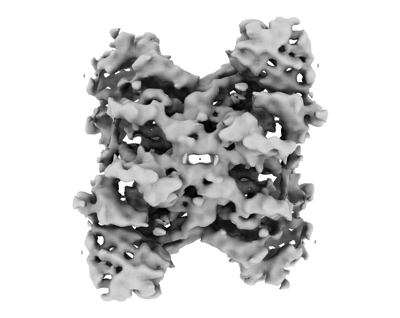 emd_24799.png emd_24799.png | 65.5 KB | ||
| Masks | emd_24799_msk_1.map | 64 MB | Mask map | |
| Others | emd_24799_half_map_1.map.gzemd_24799_half_map_2.map.gz | 48.3 MB 48.5 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-24799ftp://ftp.pdbj.org/pub/emdb/structures/EMD-24799 http://ftp.pdbj.org/pub/emdb/structures/EMD-24799ftp://ftp.pdbj.org/pub/emdb/structures/EMD-24799 | HTTPS FTP |
-Validation report
| Summary document | emd_24799_validation.pdf.gz | 500.5 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_24799_full_validation.pdf.gz | 500.1 KB | Display | |
| Data in XML | emd_24799_validation.xml.gz | 16 KB | Display | |
| Data in CIF | emd_24799_validation.cif.gz | 21 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-24799ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-24799 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10867 (Title: Rabbit Muscle Aldolase 20% glycerol, 300kV / Data size: 580.1 Data #1: Unaligned multiframe micrographs from rabbit muscle aldolase in 20% glycerol, imaged at 300kev [micrographs - multiframe]) |













-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_24799.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Rabbit muscle aldolase determined in the presence of 20% v/v glycerol. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.045 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_24799_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Rabbit muscle aldolase determined in the presence of...
| File | emd_24799_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Rabbit muscle aldolase determined in the presence of 20% v/v glycerol. Half map 1. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Rabbit muscle aldolase determined in the presence of...
| File | emd_24799_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Rabbit muscle aldolase determined in the presence of 20% v/v glycerol. Half map 2. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Aldolase
| Entire | Name: Aldolase |
|---|---|
| Components |
|
-Supramolecule #1: Aldolase
| Supramolecule | Name: Aldolase / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 157 KDa |
-Macromolecule #1: Aldolase
| Macromolecule | Name: Aldolase / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Sequence | String: PHSHPALTPE QKKELSDIAH RIVAPGKGIL AADESTGSIA KRLQSIGTEN TEENRRFYRQ LLLTADDRVN PCIGGVILFH ETLYQKADDG RPFPQVIKSK GGVVGIKVDK GVVPLAGTNG ETTTQGLDGL SERCAQYKKD GADFAKWRCV LKIGEHTPSA LAIMENANVL ...String: PHSHPALTPE QKKELSDIAH RIVAPGKGIL AADESTGSIA KRLQSIGTEN TEENRRFYRQ LLLTADDRVN PCIGGVILFH ETLYQKADDG RPFPQVIKSK GGVVGIKVDK GVVPLAGTNG ETTTQGLDGL SERCAQYKKD GADFAKWRCV LKIGEHTPSA LAIMENANVL ARYASICQQN GIVPIVEPEI LPDGDHDLKR CQYVTEKVLA AVYKALSDHH IYLEGTLLKP NMVTPGHACT QKYSHEEIAM ATVTALRRTV PPAVTGVTFL SGGQSEEEAS INLNAINKCP LLKPWALTFS YGRALQASAL KAWGGKKENL KAAQEEYVKR ALANSLACQG KYTPSGQAGA AASESLFISN HAY |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.6 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Grid | Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - Material: GOLD / Support film - topology: HOLEY ARRAY / Support film - Film thickness: 50.0 nm / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER / Pretreatment - Pressure: 9.33257 kPa | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: HOMEMADE PLUNGER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 77.0 K / Max: 77.0 K |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-50 / Number grids imaged: 1 / Number real images: 2591 / Average exposure time: 10.0 sec. / Average electron dose: 52.5 e/Å2 Details: Images were collected using stage position navigation to the center of 4 holes and then beam shift was used to target exposures. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 1.7 µm / Calibrated defocus min: 1.3 µm / Calibrated magnification: 47846 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.7 µm / Nominal defocus min: 1.3 µm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |


