 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-20786 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CYP102A1_A82F_open_state1 | |||||||||
 Map data Map data | full-length CYP102A1 in open state I | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 8.3 Å | |||||||||
 Authors Authors | Su M / Chakraborty S / Osawa Y / Zhang H | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2020 Title: Cryo-EM reveals the architecture of the dimeric cytochrome P450 CYP102A1 enzyme and conformational changes required for redox partner recognition. Authors: Min Su / Sumita Chakraborty / Yoichi Osawa / Haoming Zhang / Abstract: Cytochrome P450 family 102 subfamily A member 1 (CYP102A1) is a self-sufficient flavohemeprotein and a highly active bacterial enzyme capable of fatty acid hydroxylation at a >3,000 min turnover rate. ...Cytochrome P450 family 102 subfamily A member 1 (CYP102A1) is a self-sufficient flavohemeprotein and a highly active bacterial enzyme capable of fatty acid hydroxylation at a >3,000 min turnover rate. The CYP102A1 architecture has been postulated to be responsible for its extraordinary catalytic prowess. However, the structure of a functional full-length CYP102A1 enzyme remains to be determined. Herein, we used a cryo-EM single-particle approach, revealing that full-length CYP102A1 forms a homodimer in which both the heme and FAD domains contact each other. The FMN domain of one monomer was located close to the heme domain of the other monomer, exhibiting a configuration. Moreover, full-length CYP102A1 is highly dynamic, existing in multiple conformational states, including open and closed states. In the closed state, the FMN domain closely contacts the FAD domain, whereas in the open state, one of the FMN domains rotates away from its FAD domain and traverses to the heme domain of the other monomer. This structural arrangement and conformational dynamics may facilitate rapid intraflavin and FMN-to-heme electron transfers (ETs). Results with a variant having a 12-amino-acid deletion in the CYP102A1 linker region, connecting the catalytic heme and the diflavin reductase domains, further highlighted the importance of conformational dynamics in the ET process. Cryo-EM revealed that the Δ12 variant homodimer is conformationally more stable and incapable of FMN-to-heme ET. We conclude that closed-to-open alternation is crucial for redox partner recognition and formation of an active ET complex for CYP102A1 catalysis. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_20786.map.gz | 7.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-20786-v30.xmlemd-20786.xml | 10.4 KB 10.4 KB | Display Display | EMDB header |
| Images |  emd_20786.png emd_20786.png | 60.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20786ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20786 http://ftp.pdbj.org/pub/emdb/structures/EMD-20786ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20786 | HTTPS FTP |
-Validation report
| Summary document | emd_20786_validation.pdf.gz | 78.2 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_20786_full_validation.pdf.gz | 77.3 KB | Display | |
| Data in XML | emd_20786_validation.xml.gz | 495 B | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-20786ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-20786 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_20786.map.gz / Format: CCP4 / Size: 8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | full-length CYP102A1 in open state I | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.84 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
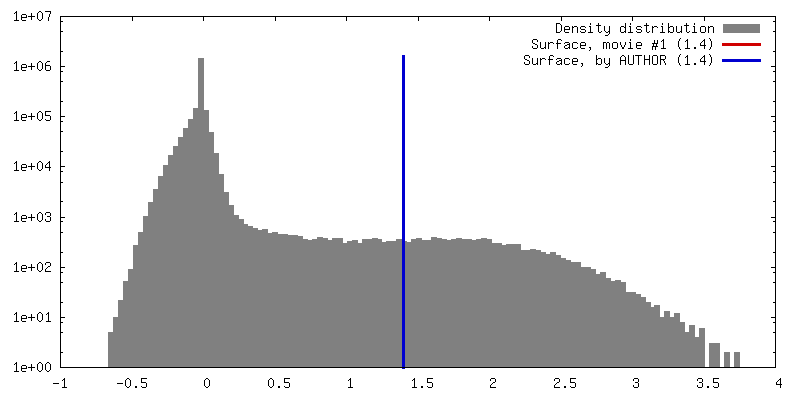
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Full-length cytochrome P450 CYP102A1 enzyme
| Entire | Name: Full-length cytochrome P450 CYP102A1 enzyme |
|---|---|
| Components |
|
-Supramolecule #1: Full-length cytochrome P450 CYP102A1 enzyme
| Supramolecule | Name: Full-length cytochrome P450 CYP102A1 enzyme / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all / Details: recombinant A82F variant |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Molecular weight | Experimental: 238.8 KDa |
-Macromolecule #1: Cytochrome P450 CYP102A1 enzyme
| Macromolecule | Name: Cytochrome P450 CYP102A1 enzyme / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Sequence | String: MHHHHHHIKE MPQPKTFGEL KNLPLLNTDK PVQALMKIAD ELGEIFKFEA PGRVTRYLSS QRLIKEACDE SRFDKNLSQA LKFVRDFFGD GLFTSWTHEK NWKKAHNILL PSFSQQAMKG YHAMMVDIAV QLVQKWERLN ADEHIEVPED MTRLTLDTIG LCGFNYRFNS ...String: MHHHHHHIKE MPQPKTFGEL KNLPLLNTDK PVQALMKIAD ELGEIFKFEA PGRVTRYLSS QRLIKEACDE SRFDKNLSQA LKFVRDFFGD GLFTSWTHEK NWKKAHNILL PSFSQQAMKG YHAMMVDIAV QLVQKWERLN ADEHIEVPED MTRLTLDTIG LCGFNYRFNS FYRDQPHPFI TSMVRALDEA MNKQQRANPD DPAYDENKRQ FQEDIKVMND LVDKIIADRK ASGEQSDDLL THMLNGKDPE TGEPLDDENI RYQIITFLIA GHETTSGLLS FALYFLVKNP HVLQKAAEEA ARVLVDPVPS YKQVKQLKYV GMVLNEALRL WPTAPAFSLY AKEDTVLGGE YPLEKGDELM VLIPQLHRDK TIWGDDVEEF RPERFENPSA IPQHAFKPFG NGQRACIGQQ FALHEATLVL GMMLKHFDFE DHTNYELDIK ETLTLKPEGF VVKAKSKKIP LGGIPSPSTE QSAKKVRKKA ENAHNTPLLV LYGSNMGTAE GTARDLADIA MSKGFAPQVA TLDSHAGNLP REGAVLIVTA SYNGHPPDNA KQFVDWLDQA SADEVKGVRY SVFGCGDKNW ATTYQKVPAF IDETLAAKGA ENIADRGEAD ASDDFEGTYE EWREHMWSDV AAYFNLDIEN SEDNKSTLSL QFVDSAADMP LAKMHGAFST NVVASKELQQ PGSARSTRHL EIELPKEASY QEGDHLGVIP RNYEGIVNRV TARFGLDASQ QIRLEAEEEK LAHLPLAKTV SVEELLQYVE LQDPVTRTQL RAMAAKTVCP PHKVELEALL EKQAYKEQVL AKRLTMLELL EKYPACEMKF SEFIALLPSI RPRYYSISSS PRVDEKQASI TVSVVSGEAW SGYGEYKGIA SNYLAELQEG DTITCFISTP QSEFTLPKDP ETPLIMVGPG TGVAPFRGFV QARKQLKEQG QSLGEAHLYF GCRSPHEDYL YQEELENAQS EGIITLHTAF SRMPNQPKTY VQHVMEQDGK KLIELLDQGA HFYICGDGSQ MAPAVEATLM KSYADVHQVS EADARLWLQQ LEEKGRYAKD VWAG |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3 mg/mL |
|---|---|
| Buffer | pH: 7.4 / Details: phosphate-buffered saline |
| Grid | Support film - Material: CARBON / Support film - topology: HOLEY / Support film - Film thickness: 0.12 nm / Details: unspecified |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK I |
| Details | The sample was monodisperse |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Average electron dose: 48.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 8.3 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 130000 |
|---|---|
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD |
