 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1run: CATABOLITE GENE ACTIVATOR PROTEIN (CAP)/DNA COMPLEX + ADENOSINE-3... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1run | ||||||
|---|---|---|---|---|---|---|---|
| Title | CATABOLITE GENE ACTIVATOR PROTEIN (CAP)/DNA COMPLEX + ADENOSINE-3',5'-CYCLIC-MONOPHOSPHATE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA /  TRANSCRIPTION REGULATION / 3D-STRUCTURE / DNA-BINDING / CAMP-BINDING / ACTIVATOR / TRANSCRIPTION-DNA COMPLEX TRANSCRIPTION REGULATION / 3D-STRUCTURE / DNA-BINDING / CAMP-BINDING / ACTIVATOR / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationcarbon catabolite repression of transcription / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / cAMP binding / protein-DNA complex / sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / DNA-templated transcription / positive regulation of DNA-templated transcription ...carbon catabolite repression of transcription / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / cAMP binding / protein-DNA complex / sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / DNA-templated transcription / positive regulation of DNA-templated transcription / identical protein binding / cytosolSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.7 Å | ||||||
 Authors Authors | Parkinson, G.N. / Gunasekera, A. / Vojtechovsky, J. / Zhang, X. / Kunkel, T.A. / Berman, H.M. / Ebright, R.H. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1996 Title: Aromatic hydrogen bond in sequence-specific protein DNA recognition. Authors: Parkinson, G. / Gunasekera, A. / Vojtechovsky, J. / Zhang, X. / Kunkel, T.A. / Berman, H. / Ebright, R.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1run.cif.gz | 131.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1run.ent.gz | 99.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1run.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ru/1runftp://data.pdbj.org/pub/pdb/validation_reports/ru/1run | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj








































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 4352.867 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: DNA chain | Mass: 5152.358 Da / Num. of mol.: 2 / Source method: obtained synthetically #3: Protein | Mass: 23541.242 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Production host: Escherichia coli (E. coli) / References: UniProt: P0ACJ8#4: Chemical | Cyclic adenosine monophosphate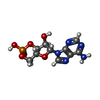  Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 201 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 201 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 6 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.5 Å3/Da / Density % sol: 58.68 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 258 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Beamline: F1 |
| Detector | Type: FUJI / Detector: IMAGE PLATE / Date: Sep 1, 1993 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.7→40 Å / Num. obs: 17777 / % possible obs: 83 % / Observed criterion σ(I): 2 / Redundancy: 2.7 % / Rmerge(I) obs: 0.116 |
| Reflection | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 40 Å / % possible obs: 83 % / Observed criterion σ(I): 2 / Redundancy: 2.7 % / Num. measured all: 48657 |
- Processing
Processing
| Software |
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.7→10 Å / σ(F): 4
| ||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→10 Å
| ||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||
| Refinement | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 10 Å / σ(F): 4 / Rfactor obs: 0.207 | ||||||||||||
| Solvent computation | *PLUS | ||||||||||||
| Displacement parameters | *PLUS Biso mean: 26.7 Å2 | ||||||||||||
| Refine LS restraints | *PLUS
|
