ジャーナル: Acta Crystallogr D Struct Biol / 年: 2019 タイトル: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. 著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / ...著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams / 要旨: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報
 マップデータ
マップデータ 試料
試料 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) データ登録者
データ登録者 ポーランド, 1件
ポーランド, 1件  引用
引用


 構造の表示
構造の表示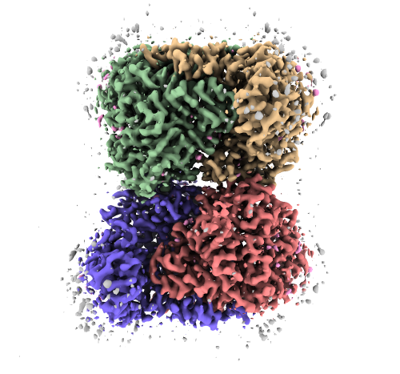
 ダウンロードとリンク
ダウンロードとリンク emd_18436.png
emd_18436.png http://ftp.pdbj.org/pub/emdb/structures/EMD-18436
http://ftp.pdbj.org/pub/emdb/structures/EMD-18436


 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















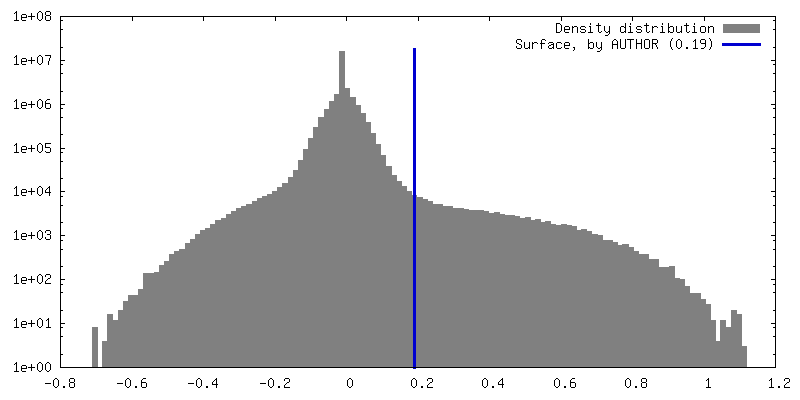










 試料の構成要素
試料の構成要素

 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN