 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-16483: In vitro structure of the Nitrosopumilus maritimus S-layer - Two-... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | In vitro structure of the Nitrosopumilus maritimus S-layer - Two-fold symmetry (C2) | |||||||||
 Map data Map data | PostProcessed map with B-factor sharpening | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Nmar_1547 S-layer /  STRUCTURAL PROTEIN STRUCTURAL PROTEIN | |||||||||
| Function / homology | membrane / Uncharacterized protein Function and homology information Function and homology information | |||||||||
| Biological species |  Nitrosopumilus maritimus SCM1 (archaea) Nitrosopumilus maritimus SCM1 (archaea) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.71 Å | |||||||||
 Authors Authors | von Kuegelgen A / Bharat T | |||||||||
| Funding support |  United Kingdom, 2 items United Kingdom, 2 items
| |||||||||
 Citation Citation | Journal: To Be Published Title: Membrane-less channels sieve cations in ammonia-oxidising marine archaea Authors: von Kuegelgen A / Cassidy CK / van Dorst S / Pagani LL / Ford Z / Loewe J / Stansfeld PJ / Bharat TAM | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
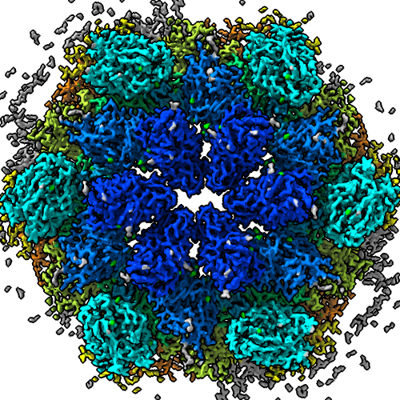
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_16483.map.gz | 114 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-16483-v30.xmlemd-16483.xml | 26.2 KB 26.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_16483_fsc.xml | 22.4 KB | Display | FSC data file |
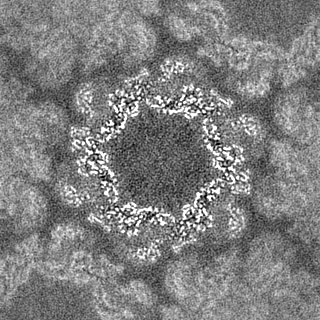
| Images |  emd_16483.png emd_16483.png | 290 KB | ||
| Masks | emd_16483_msk_1.map | 125 MB | Mask map | |
| Filedesc metadata | emd-16483.cif.gz | 8.6 KB | ||
| Others | emd_16483_additional_1.map.gzemd_16483_half_map_1.map.gzemd_16483_half_map_2.map.gz | 112.5 MB 113.7 MB 113.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-16483ftp://ftp.pdbj.org/pub/emdb/structures/EMD-16483 http://ftp.pdbj.org/pub/emdb/structures/EMD-16483ftp://ftp.pdbj.org/pub/emdb/structures/EMD-16483 | HTTPS FTP |
-Related structure data
| Related structure data |  8c8lMC  8c8kC  8c8mC  8c8nC  8c8oC  8c8rC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_16483.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | PostProcessed map with B-factor sharpening | ||||||||||||||||||||
| Voxel size | X=Y=Z: 1.092 Å | ||||||||||||||||||||
| Density |
| ||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||
| Details | EMDB XML:
|
-Supplemental data
-Mask #1
| File | emd_16483_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
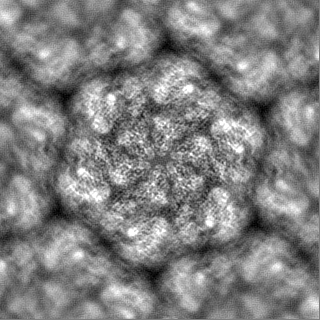
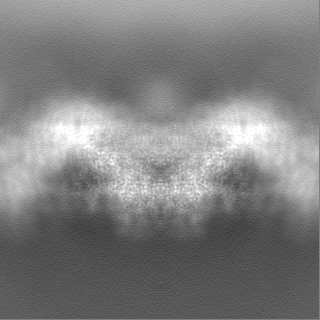
| Projections & Slices |
| ||||||||||||
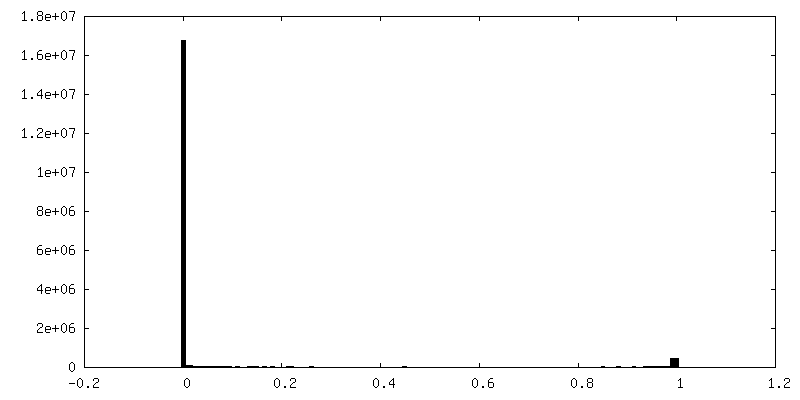
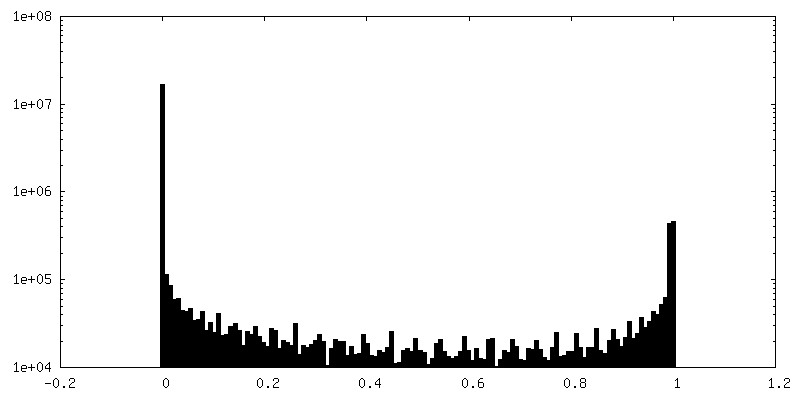
| Density Histograms |
 Z
Z Y
Y X
X







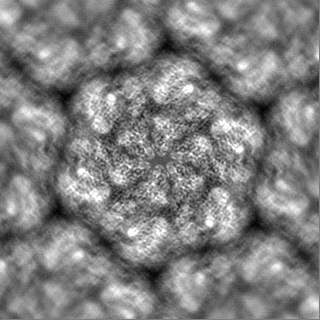
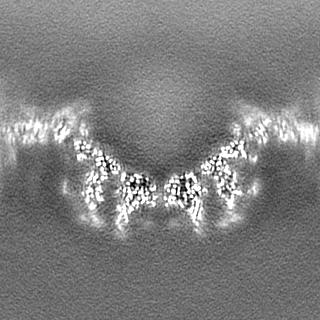
-Additional map: Full map with out b-factor sharpening
| File | emd_16483_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Full map with out b-factor sharpening | ||||||||||||
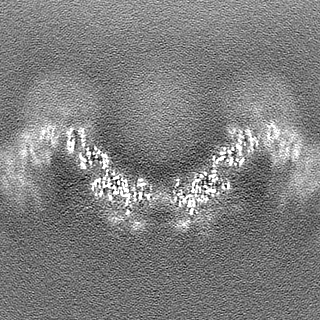
| Projections & Slices |
| ||||||||||||
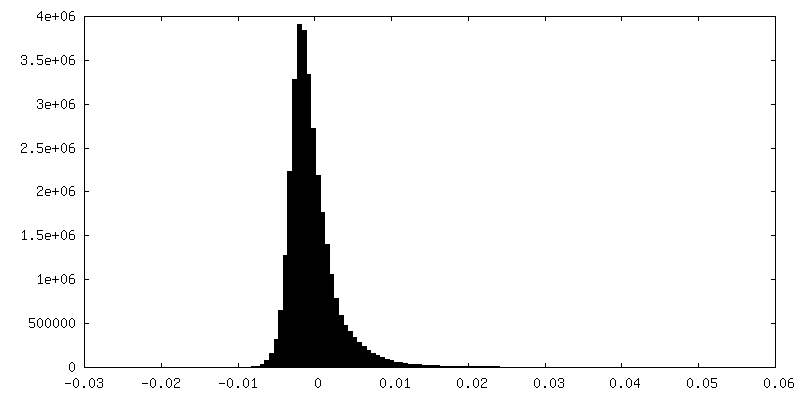
| Density Histograms |





-Half map: Half map 2
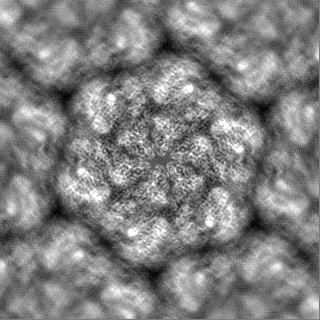
| File | emd_16483_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2 | ||||||||||||
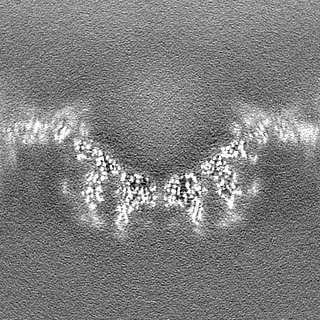
| Projections & Slices |
| ||||||||||||
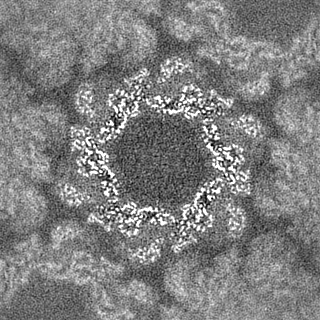
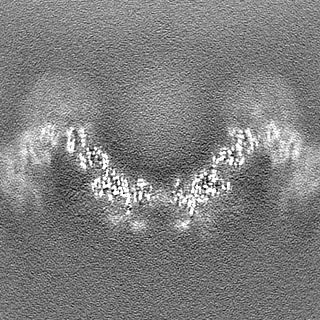
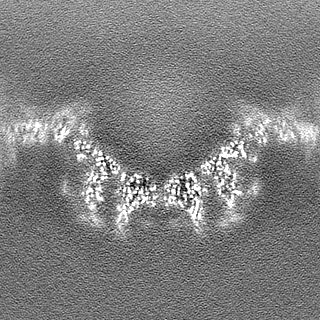
| Density Histograms |

-Half map: Half map 1
| File | emd_16483_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

- Sample components
Sample components
-Entire : Nitrosopumilus maritimus S-layer
| Entire | Name: Nitrosopumilus maritimus S-layer |
|---|---|
| Components |
|
-Supramolecule #1: Nitrosopumilus maritimus S-layer
| Supramolecule | Name: Nitrosopumilus maritimus S-layer / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all / Details: Nitrosopumilus maritimus S-layer C2 symmetrised |
|---|---|
| Source (natural) | Organism: Nitrosopumilus maritimus SCM1 (archaea) / Strain: SCM1 / Location in cell: extracellular |
-Macromolecule #1: Cell surface protein
| Macromolecule | Name: Cell surface protein / type: protein_or_peptide / ID: 1 / Details: In-vitro isolated S-layer / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Nitrosopumilus maritimus SCM1 (archaea) / Strain: SCM1 |
| Molecular weight | Theoretical: 183.156 KDa |
| Sequence | String: MNNEIGRKIT SLTLMTIMVA GGLTFAIPGV MPEAMAANAN LFVSAENSQF DNYMSGPQVI EVVVIDSDIN DTDEAKGEPD VTVNGKVLR MVQAVDGNWY GYFADRDQAQ IADSTATTAD SGLDFGVFCA SSSGTAALGF STTETDGIAI PITIANATAT G NGTQTGSS ...String: MNNEIGRKIT SLTLMTIMVA GGLTFAIPGV MPEAMAANAN LFVSAENSQF DNYMSGPQVI EVVVIDSDIN DTDEAKGEPD VTVNGKVLR MVQAVDGNWY GYFADRDQAQ IADSTATTAD SGLDFGVFCA SSSGTAALGF STTETDGIAI PITIANATAT G NGTQTGSS SGGAITTTCA ANTLDASTAN GTINVVREAK DPVAASGSVS VGQIGLKNGT ANSGPNWPFI QLYELNPTGN VV VQYNKGG GVQSTTLTFD TVDQFAELSL DRTVFPRVSQ VHATITDLWL NIDPTDEDSW TFATNTKNTT SSFNVDTFYQ VFD ENGASG GSALTLRTTL SSLMCEDNCV LTLDVDAQSS GTPVVTIQDN GDSILTQLNA SSNTNANNAS AFGISTETAK LGTG SIPVT ITEQGPNSGV FGTYDESDKS VLKITDNAKR GTSASLDYNE TPQTILVGFS FASIDIQPVT DEWTSGQEIP VVIVD ADQN KNSRADEDLD LNNPDVTLIP ALRTGDPFTI DEGGTPSLIF TNGTNGDDSI FDTGAINNTS AGQVGNFTLN INVTRF SSA TNITSTESID TFSKRLISAQ TANSSANFDV DFAIIDLGSA TLETLKETVV DEDNTAVGFN FFNYDVRSLG ADTVSIA LL NTTGNILPWV NNDTRNVDKN NAILLVSNST NSQAYVDLTN AVSDAVYGST NTDSNVNIGF AMYFTGVGDL AAKEVIVM D FFSFGFTDDG VQSSERFANQ IIRIEAEETG DNTSTFEGSL EYVMVNQINI QDAGTFSGIT PIADDPSFIV IEDLTDEDA PRVNYNDLGA DGVTTPVSDQ EEAPSHSGVV SLNADSYKIA DTVVITVEDL DLNVDSDLID IFTVVSDNSK ATDDAVGSAT TQSLSFGEL GRLLDVTFDD VIWSTPDGAN NTATGNDSDT CSTELSNAGI TDTGLGATGF TLVETGAATG VFVGDFQIPS F WCRVSDTT TTPYTYAGDE ETTTGLDIEV NYVDFRDASG EIVEVGDSAG VRANTGSVSL DRTVYPVPFG TIADSSKAAN AA PNGRSVF PIHATGITST IDSTEELPTG DLTIHVRIND PDFDENPAGE DAMDQDNALK ISVIRGSDSV VLGYAGASER TGK IDVGGN NGTISNIRSF GEMDEIAPDA GIFELDVNIK FTDGPASAQC NSHDTLYTAL DGTTGKADTN RFDDGAASGQ EYCI LQGDI LQVEYTDPAD ASGDANTVTD SATFDLRNGV LQSDKSVYII GSDMILTLIE PDFDLDNDSA ETYDLDLIEW DSDAA TTTM GNKGVTGAAA AFDPEPTDFR ETGDSTGIFQ IVIEIPESLS NDKLERGEEI ILEYTDWGPS GSDYVGDEDE DVNLTI YTS NFGATVELDQ KVYSWTDKVY ITIVAPDHNF DSDLVDEIGE TDSDPIKVST RGFDLDNYKL VETGTDTGIF TGEVILT GF TAHDADGDGN TGDATGTTSG SGPTDGLLAT DDDDGLTVSF EFSEDETIVG SALIRWNIGE VQWLEASYPA SGTGVVRV I DPDMNLDPEA VDNFEVDVWS DSDAGGIDLT VTETNEATGI FEGTVFFTTL DESSGHRLRV SEGDTVTAEY EDNTLPDPY TTADELDITA TSLIGTVVPP LERAPAANLR TVDAFGNSLD SVSVDQQVQI SADLANGQDR EQSFAYLVQI QDANGVTVSL AWITGSLSS GQSFSPALSW IPTEAGTYTA TAFVWESVDN PTALSPPVST TVNVS UniProtKB: Uncharacterized protein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | 2D array |
-Sample preparation
| Buffer | pH: 7.5 Component:
Details: 50 mM HEPES/NaOH pH=7.5, 500 mM NaCl, 50 mM MgCl2, 10 mM CaCl2 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grid | Model: Quantifoil R2/2 / Material: COPPER/RHODIUM / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 20 sec. / Pretreatment - Atmosphere: AIR / Details: 15 mA | |||||||||||||||
| Vitrification | Cryogen name: NITROGEN / Chamber humidity: 100 % / Chamber temperature: 283.15 K / Instrument: FEI VITROBOT MARK IV Details: absorption for 60 sec and blotted for 5 sec with blot force -10. | |||||||||||||||
| Details | In vitro isolate S-layer cell envelopes |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 5.0 µm / Calibrated defocus min: 2.0 µm / Calibrated magnification: 81000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 5.0 µm / Nominal defocus min: 2.0 µm / Nominal magnification: 81000 |
| Specialist optics | Spherical aberration corrector: not used / Chromatic aberration corrector: not used / Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 3 / Number real images: 12557 / Average exposure time: 4.2 sec. / Average electron dose: 48.5 e/Å2 Details: collected over three sessions with one session with the stage tilted by 33 degrees (alpha tilt) |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 1971908 Details: Initially, side views of S-layer sheets were first manually picked along the edge of the lattice using the helical picking tab in RELION while setting the helical rise to 60 angstrom. Top ...Details: Initially, side views of S-layer sheets were first manually picked along the edge of the lattice using the helical picking tab in RELION while setting the helical rise to 60 angstrom. Top and tilted views were manually picked at the central hexameric axis. Manually picked particles were extracted in 4x downsampled 128x128 pixel2 boxes and classified using reference-free 2D classification inside RELION-3.1. Class averages centered at a hexameric axis were used to automatically pick particles inside RELION-3.1. Automatically picked particles were extracted in 4x downsampled 128x128 pixel2 boxes and classified using reference-free 2D classification. Particle coordinates belonging to class averages centered at the hexameric axis were used to train TOPAZ in 5x downsampled micrographs with the neural network architecture conv127. For the final reconstruction, particles were picked using TOPAZ and the previously trained neural network above. Additionally, top, bottom, and side views were picked using the reference-based autopicker inside RELION-3.1, which TOPAZ did not readily identify. Particles were extracted in 4x downsampled 128x128 pixel2 boxes and classified using reference-free 2D classification inside RELION-3.1. Particles belonging to class averages centered at the hexameric axis were combined, and particles within 30 angstrom were removed to prevent duplication after alignment. All resulting particles were then re-extracted in 4x downsampled 128x128 pixel2 boxes. |
|---|---|
| Startup model | Type of model: NONE Details: All side views and a subset of top and bottom views were used for initial model generation in RELION-3.1. The scaled and lowpass filtered output was then used as a starting model for 3D auto ...Details: All side views and a subset of top and bottom views were used for initial model generation in RELION-3.1. The scaled and lowpass filtered output was then used as a starting model for 3D auto refinement in a 512x512 pixel box. |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) / Details: Angle assignment was performed within RELION3.1 |
| Final 3D classification | Software - Name: RELION (ver. 3.1) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) / Details: Angle assignment was performed within RELION3.1 |
| Final reconstruction | Applied symmetry - Point group: C2 (2 fold cyclic) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 2.71 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.1) Details: Per-particle defocus, anisotropy magnification, and higher-order aberrations were refined inside RELION3.1, followed by three rounds of focused 3D auto refinement. Bayesian particle ...Details: Per-particle defocus, anisotropy magnification, and higher-order aberrations were refined inside RELION3.1, followed by three rounds of focused 3D auto refinement. Bayesian particle polishing was performed subsequently in a 640x640 pixel2 box followed by auto-refinement and symmetry relaxation. The final map was obtained from 354,860 particles and post-processed using a soft mask focused on the central hexamer, yielding a global resolution of 2.7 angstrom according to the Fourier shell correlation criterion between two independently refined half-maps at a threshold value at 0.143, and local resolution up to 2.5 angstrom Number images used: 354860 |
| Details | Movies collected at the scope were clustered into optics groups based on the XML meta-data of the data-collection software EPU (Thermo Fisher Scientific) using a k-means algorithm implemented in EPU_group_AFIS (https://github.com/DustinMorado/EPU_group_AFIS). Imported movies were motion-corrected, dose-weighted, and Fourier cropped (2x) with MotionCor2 implemented in RELION-3.1. Contrast transfer functions (CTFs) of the resulting motion-corrected micrographs were estimated using CTFFIND4. |
| FSC plot (resolution estimation) | 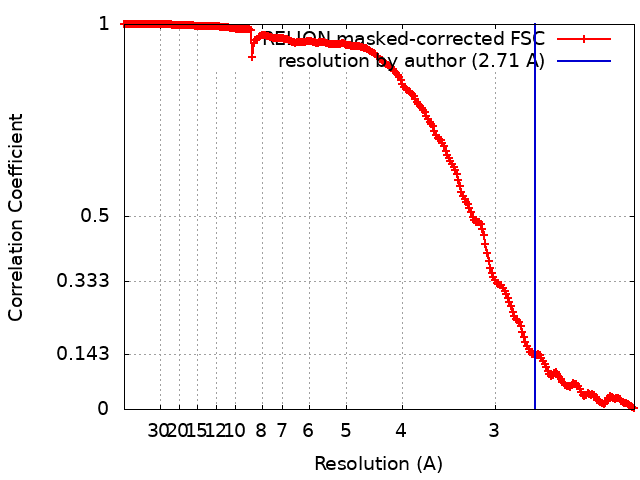 |
-Atomic model buiding 1
| Refinement | Space: RECIPROCAL / Protocol: AB INITIO MODEL / Overall B value: 41.4 / Target criteria: Best Fit |
|---|---|
| Output model | PDB-8c8l: |
