 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1502: Initial location of the RNA-dependent RNA polymerase in the bacte... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1502 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Initial location of the RNA-dependent RNA polymerase in the bacteriophage phi6 procapsid determined by cryo-electron microscopy | |||||||||
 Map data Map data | Map of a procapsid without the P2 protein, containing only proteins P1, P4 and P7 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Cystoviridae / P2 protein | |||||||||
| Biological species |  Pseudomonas phage phi6 (bacteriophage) Pseudomonas phage phi6 (bacteriophage) | |||||||||
| Method | single particle reconstruction / cryo EM / negative staining / Resolution: 11.0 Å | |||||||||
 Authors Authors | Sen A / Heymann JB / Cheng N / Qiao J / Mindich L / Steven AC | |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2008 Title: Initial location of the RNA-dependent RNA polymerase in the bacteriophage Phi6 procapsid determined by cryo-electron microscopy. Authors: Anindito Sen / J Bernard Heymann / Naiqian Cheng / Jian Qiao / Leonard Mindich / Alasdair C Steven /  Abstract: The RNA-dependent RNA polymerases (RdRPs) of Cystoviridae bacteriophages, like those of eukaryotic viruses of the Reoviridae, function inside the inner capsid shell in both replication and ...The RNA-dependent RNA polymerases (RdRPs) of Cystoviridae bacteriophages, like those of eukaryotic viruses of the Reoviridae, function inside the inner capsid shell in both replication and transcription. In bacteriophage Phi6, this inner shell is first assembled as an icosahedral procapsid with recessed 5-fold vertices that subsequently undergoes major structural changes during maturation. The tripartite genome is packaged as single-stranded RNA molecules via channels on the 5-fold vertices, and transcripts probably exit the mature capsid by the same route. The RdRP (protein P2) is assembled within the procapsid, and it was thought that it should be located on the 5-fold axes near the RNA entry and exit channels. To determine the initial location of the RdRP inside the procapsid of bacteriophage Phi6, we performed cryo-electron microscopy of wild type and mutant procapsids and complemented these data with biochemical determinations of copy numbers. We observe ring-like densities on the 3-fold axes that are strong in a mutant that has approximately 10 copies of P2 per particle; faint in wild type, reflecting the lower copy number of approximately 3; and completely absent in a P2-null mutant. The dimensions and shapes of these densities match those of the known crystal structure of the P2 monomer. We propose that, during maturation, the P2 molecules rotate to occupy positions closer to adjacent 5-fold vertices where they conduct replication and transcription. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
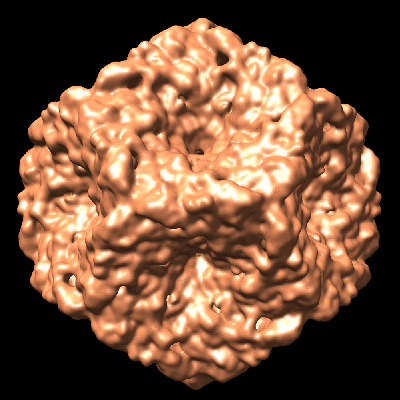
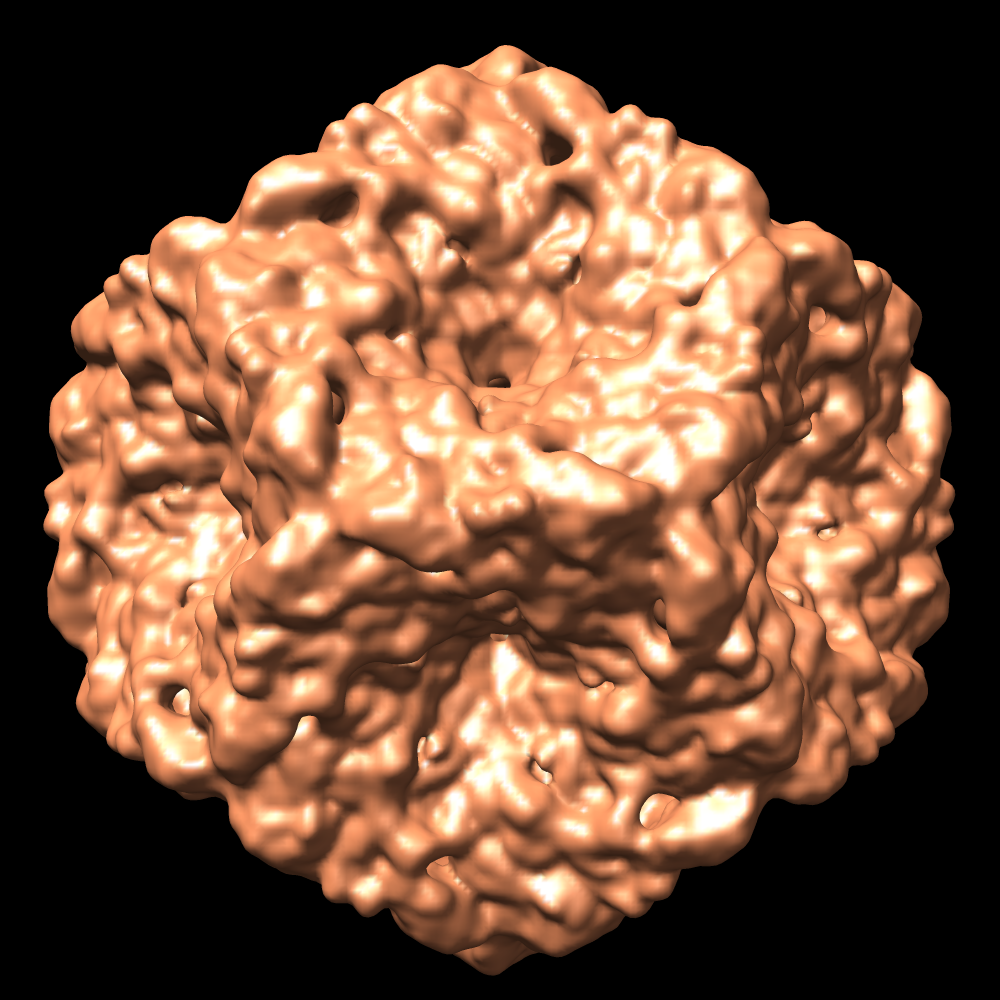

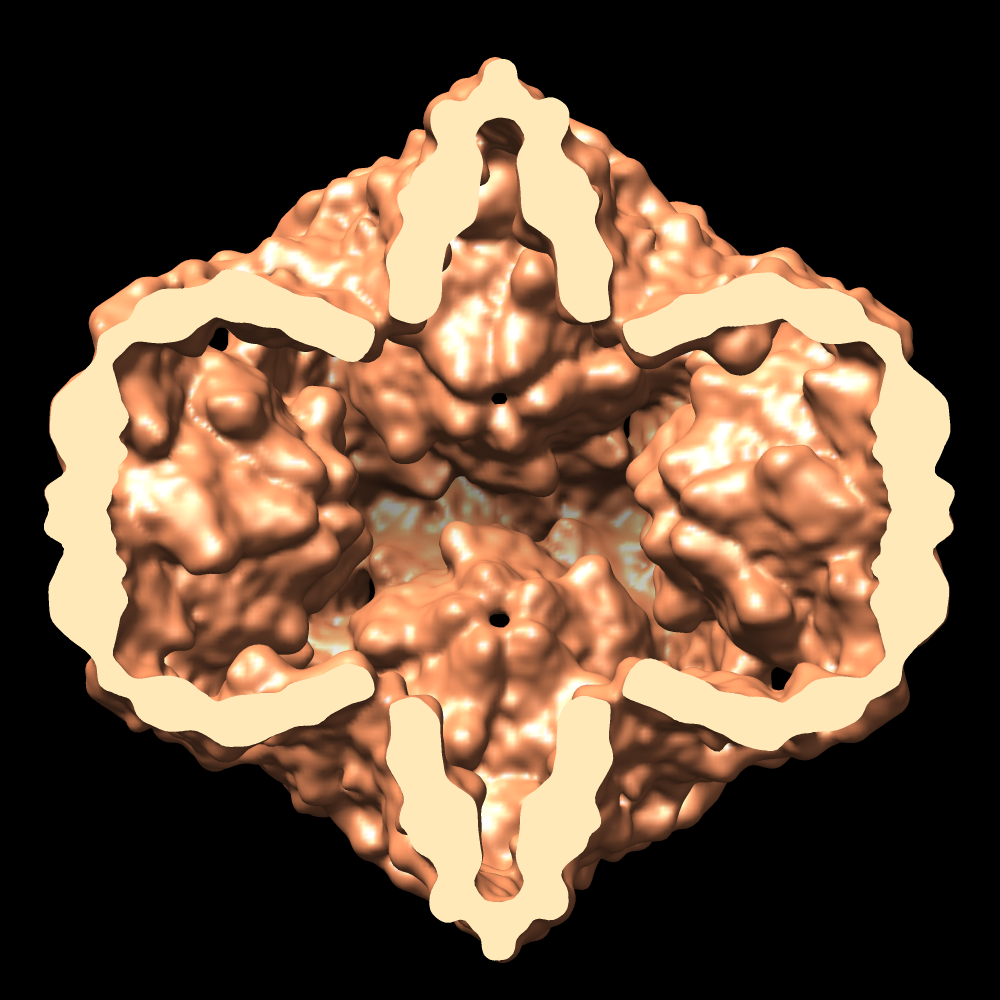
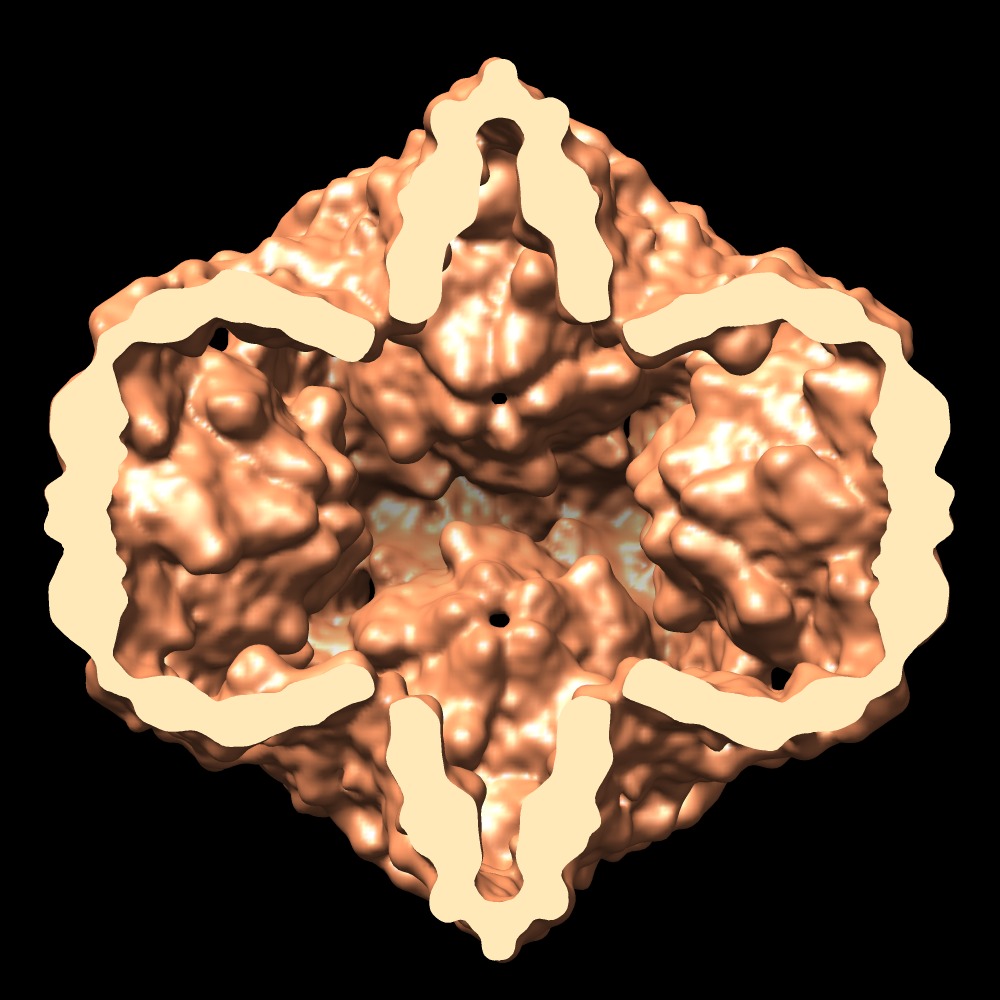
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1502.map.gz | 36.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1502-v30.xmlemd-1502.xml | 9.3 KB 9.3 KB | Display Display | EMDB header |
| Images | 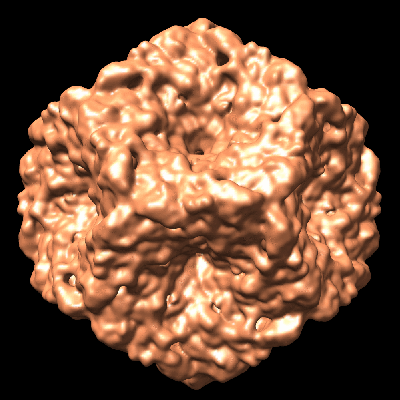 1502.gif 1502.gif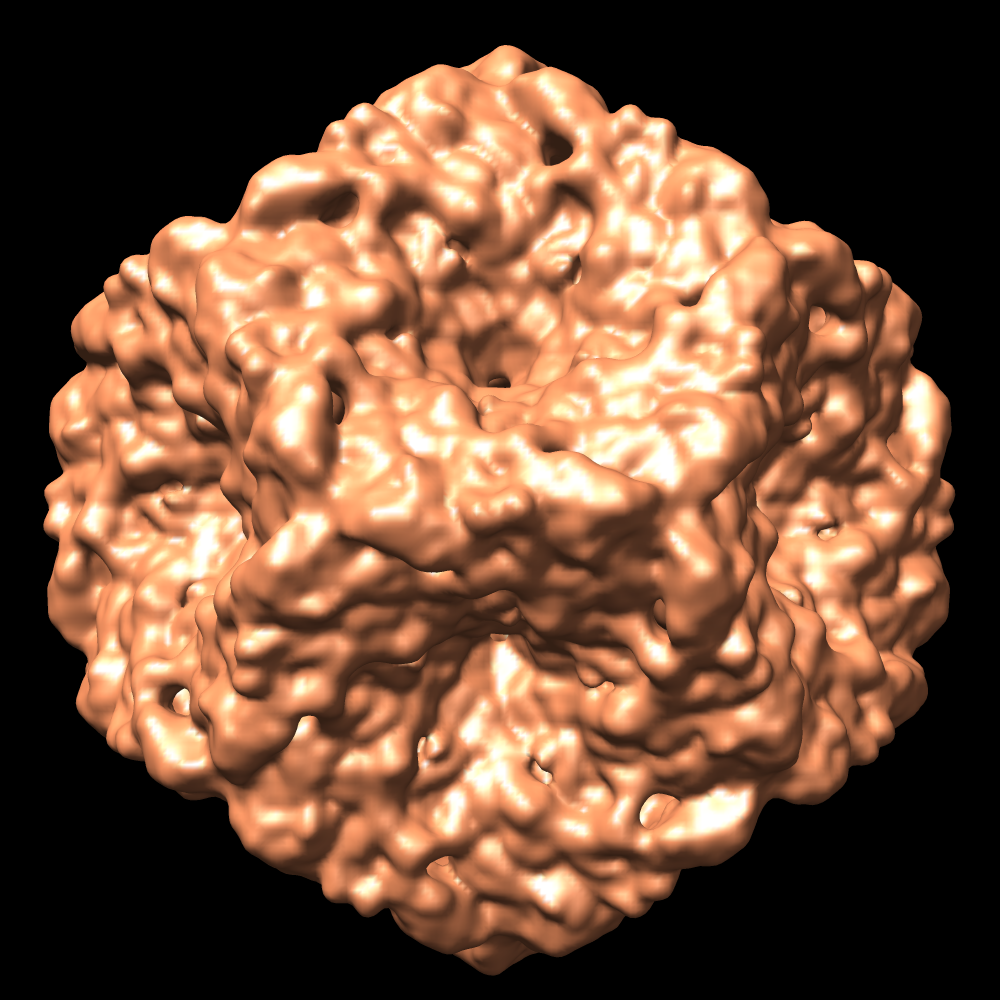 emd_1502.pngemd_1502.tif emd_1502.pngemd_1502.tif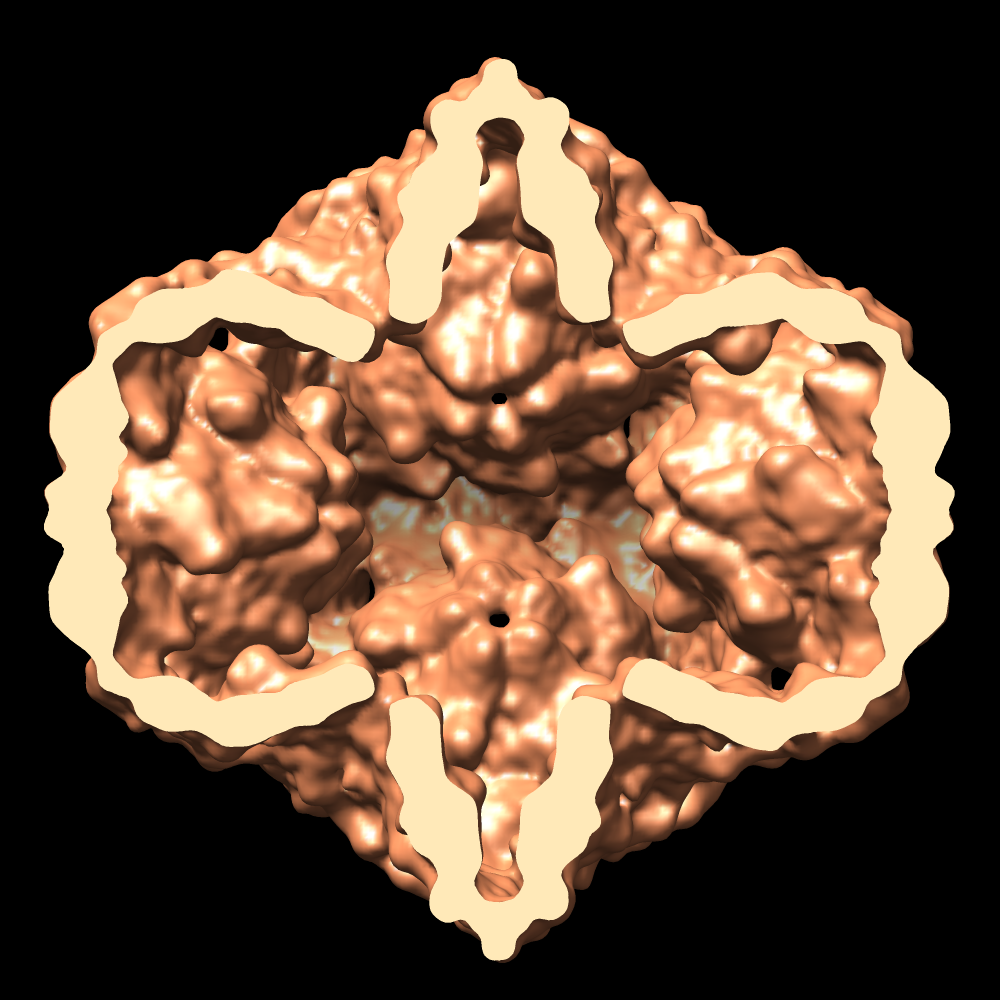 emd_1502_1.pngemd_1502_1.tif emd_1502_1.pngemd_1502_1.tif | 90.7 KB 698.2 KB 755 KB 514.3 KB 549 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1502ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1502 http://ftp.pdbj.org/pub/emdb/structures/EMD-1502ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1502 | HTTPS FTP |
-Validation report
| Summary document | emd_1502_validation.pdf.gz | 247 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_1502_full_validation.pdf.gz | 246.2 KB | Display | |
| Data in XML | emd_1502_validation.xml.gz | 6.6 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1502ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1502 | HTTPS FTP |
-Related structure data







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1502.map.gz / Format: CCP4 / Size: 47.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
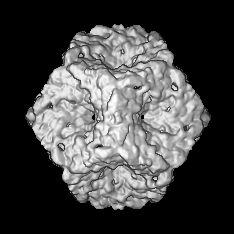
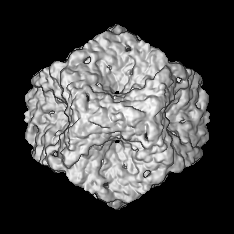
| Annotation | Map of a procapsid without the P2 protein, containing only proteins P1, P4 and P7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.54 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
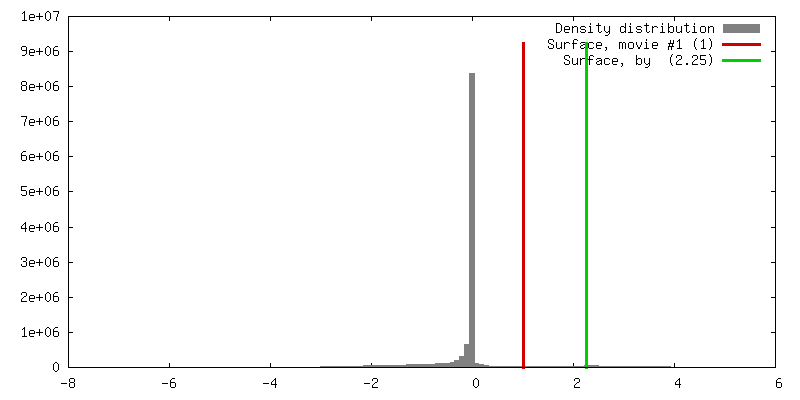
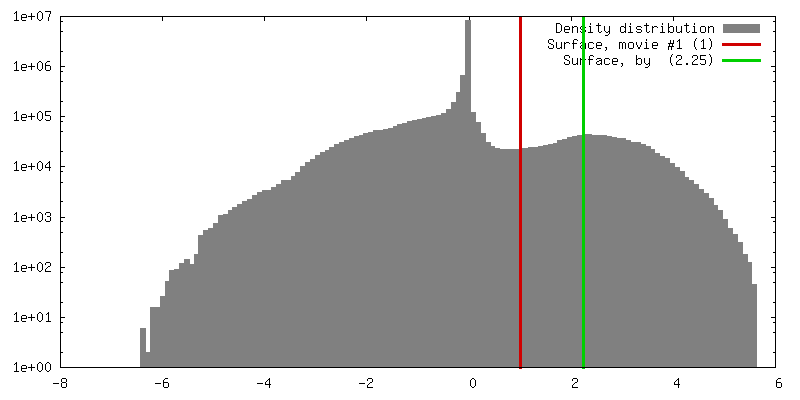
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
















-Supplemental data
- Sample components
Sample components
-Entire : Phi 6 procapsid without the P2 protein
| Entire | Name: Phi 6 procapsid without the P2 protein |
|---|---|
| Components |
|
-Supramolecule #1000: Phi 6 procapsid without the P2 protein
| Supramolecule | Name: Phi 6 procapsid without the P2 protein / type: sample / ID: 1000 Details: Produced using the Escherichia coli cell line LM4419 with the plasmid, pLM1906 Oligomeric state: Procapsid with 3 proteins P1, P4, P7 / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 14 MDa |
-Supramolecule #1: Pseudomonas phage phi6
| Supramolecule | Name: Pseudomonas phage phi6 / type: virus / ID: 1 / NCBI-ID: 10879 / Sci species name: Pseudomonas phage phi6 / Virus type: VIRION / Virus isolate: STRAIN / Virus enveloped: Yes / Virus empty: Yes |
|---|---|
| Host (natural) | Organism:  Pseudomonas syringae (bacteria) / synonym: BACTERIA(EUBACTERIA) Pseudomonas syringae (bacteria) / synonym: BACTERIA(EUBACTERIA) |
| Molecular weight | Theoretical: 14 MDa |
| Virus shell | Shell ID: 1 / Name: Procapsid / Diameter: 470 Å / T number (triangulation number): 1 |
-Experimental details
-Structure determination
| Method | negative staining, cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Details: 10mM sodium phosphate, 1mM MgCl2, 150 mM NaCl |
| Staining | Type: NEGATIVE / Details: None |
| Vitrification | Cryogen name: ETHANE / Instrument: REICHERT-JUNG PLUNGER Details: Vitrification instrument: Reichert-Jung cryo-fixation unit |
- Electron microscopy
Electron microscopy
| Microscope | FEI/PHILIPS CM200FEG |
|---|---|
| Alignment procedure | Legacy - Astigmatism: Corrected at 300000 times magnification |
| Date | Mar 15, 2007 |
| Image recording | Digitization - Scanner: NIKON SUPER COOLSCAN 9000 / Digitization - Sampling interval: 6.35 µm / Number real images: 36 / Average electron dose: 15 e/Å2 / Bits/pixel: 16 |
| Tilt angle min | 0 |
| Tilt angle max | 0 |
| Electron beam | Acceleration voltage: 120 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2 mm / Nominal defocus max: 2.3 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 50000 |
| Sample stage | Specimen holder: Side entry, Eucentric / Specimen holder model: GATAN LIQUID NITROGEN |
-Image processing
| CTF correction | Details: Each particle phase flipped |
|---|---|
| Final reconstruction | Applied symmetry - Point group: I (icosahedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 11.0 Å / Resolution method: FSC 0.33 CUT-OFF / Software - Name: Bsoft, PFT3DR / Number images used: 1072 |
