 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-14352: Structure of the GroEL chaperonin in complex with the CnoX chaper... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the GroEL chaperonin in complex with the CnoX chaperedoxin | |||||||||
 Map data Map data | Map generated by phenix.resolve after relion refinement | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Protein Folding / Redox / Complex / Chaperonin / CHAPERONE | |||||||||
| Function / homology | : / :  Function and homology information Function and homology information | |||||||||
| Biological species |  | |||||||||
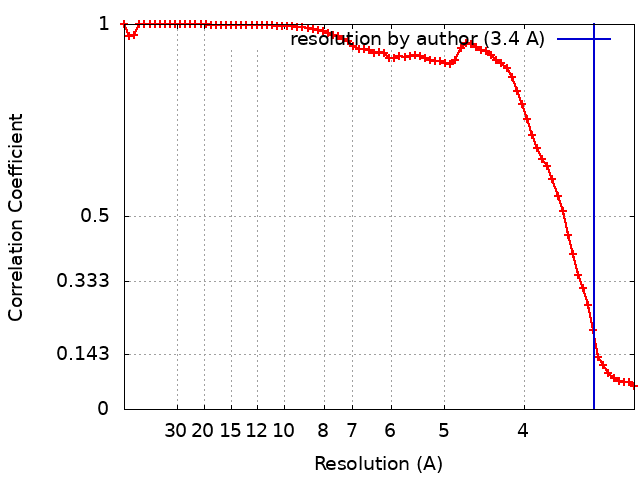
| Method | single particle reconstruction / cryo EM / Resolution: 3.4 Å | |||||||||
 Authors Authors | Van der Verren SE / Remaut H | |||||||||
| Funding support |  Belgium, 2 items Belgium, 2 items
| |||||||||
 Citation Citation | Journal: To Be Published Title: A molecular plugin rescues GroEL/ES substrates from pre-folding oxidation Authors: Dupuy E / Van der Verren SE / Lin J / Wilson M / Viela F / Latour E / Dachsbeck A / Gennaris A / Vertommen D / Dufresne Y / Iorga B / Goemans CV / Remaut H / Collet JF | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
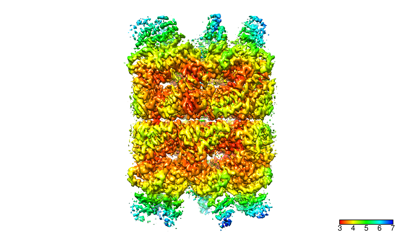
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_14352.map.gz | 7.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-14352-v30.xmlemd-14352.xml | 15.4 KB 15.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_14352_fsc.xml | 9.1 KB | Display | FSC data file |
| Images |  emd_14352.png emd_14352.png | 75.6 KB | ||
| Filedesc metadata | emd-14352.cif.gz | 5.8 KB | ||
| Others | emd_14352_additional_1.map.gz | 9.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-14352ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14352 http://ftp.pdbj.org/pub/emdb/structures/EMD-14352ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14352 | HTTPS FTP |
-Validation report
| Summary document | emd_14352_validation.pdf.gz | 480.9 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_14352_full_validation.pdf.gz | 480.5 KB | Display | |
| Data in XML | emd_14352_validation.xml.gz | 9.8 KB | Display | |
| Data in CIF | emd_14352_validation.cif.gz | 12.7 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-14352ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-14352 | HTTPS FTP |
-Related structure data
| Related structure data |  7ywyMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_14352.map.gz / Format: CCP4 / Size: 30.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Map generated by phenix.resolve after relion refinement | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.568 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Additional map: Local sharpened map as generated by LocScale to help visualise CnoX
| File | emd_14352_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Local sharpened map as generated by LocScale to help visualise CnoX | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : 1:1 stoichiometric complex of GroEL-CnoX
| Entire | Name: 1:1 stoichiometric complex of GroEL-CnoX |
|---|---|
| Components |
|
-Supramolecule #1: 1:1 stoichiometric complex of GroEL-CnoX
| Supramolecule | Name: 1:1 stoichiometric complex of GroEL-CnoX / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Chaperedoxin
| Macromolecule | Name: Chaperedoxin / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 9.771923 KDa |
| Recombinant expression | Organism: |
| Sequence | String: ADTPEIQQLQ QQVAENPEDA ALATQLALQL HQVGRNEEAL ELLFGHLRKD LTAADGQTRK TFQEILAALG TGDALASKYR RQLYALLY UniProtKB: UNIPROTKB: A0A7U2VUH6 |
-Macromolecule #2: Chaperonin GroEL
| Macromolecule | Name: Chaperonin GroEL / type: protein_or_peptide / ID: 2 / Number of copies: 14 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 55.220105 KDa |
| Recombinant expression | Organism: |
| Sequence | String: AAKDVKFGND ARVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTAAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV ...String: AAKDVKFGND ARVKMLRGVN VLADAVKVTL GPKGRNVVLD KSFGAPTITK DGVSVAREIE LEDKFENMGA QMVKEVASKA NDAAGDGTT TATVLAQAII TEGLKAVAAG MNPMDLKRGI DKAVTAAVEE LKALSVPCSD SKAIAQVGTI SANSDETVGK L IAEAMDKV GKEGVITVED GTGLQDELDV VEGMQFDRGY LSPYFINKPE TGAVELESPF ILLADKKISN IREMLPVLEA VA KAGKPLL IIAEDVEGEA LATLVVNTMR GIVKVAAVKA PGFGDRRKAM LQDIATLTGG TVISEEIGME LEKATLEDLG QAK RVVINK DTTTIIDGVG EEAAIQGRVA QIRQQIEEAT SDYDREKLQE RVAKLAGGVA VIKVGAATEV EMKEKKARVE DALH ATRAA VEEGVVAGGG VALIRVASKL ADLRGQNEDQ NVGIKVALRA MEAPLRQIVL NCGEEPSVVA NTVKGGDGNY GYNAA TEEY GNMIDMGILD PTKVTRSALQ YAASVAGLMI TTECMVTDLP UniProtKB: UNIPROTKB: A0A828EVF1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.4 mg/mL |
|---|---|
| Buffer | pH: 8 / Details: 50mM Tris pH=8, 150mM NaCl, 1mM EDTA |
| Vitrification | Cryogen name: ETHANE / Instrument: GATAN CRYOPLUNGE 3 |
| Details | Strep-affinity purified complex |
- Electron microscopy
Electron microscopy
| Microscope | JEOL CRYO ARM 300 |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 68.3 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: OTHER / Imaging mode: OTHER / Nominal defocus max: 3.0 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 60000 |
+Image processing
-Atomic model buiding 1
| Initial model |
| ||||||
|---|---|---|---|---|---|---|---|
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Correlation coeficient | ||||||
| Output model | PDB-7ywy: |


